KEY POINTS
Sickle cell disease causes a chronic hemolytic anemia associated with acute and chronic vaso-occlusion.
Baseline hemodynamic and laboratory values in patients with sickle cell disease can be confused with sepsis.
Serum creatinine levels of 1 to 1.5 mg/dL often indicate significant renal dysfunction.
The most common intensive care management problems in patients with sickle cell disease include the acute chest syndrome, very severe anemia, sepsis, stroke, priapism, splenic sequestration, or right heart failure associated with acute chest syndrome and/or acute severe hemolytic anemia.
The acute chest syndrome is a form of acute lung injury that occurs in 10% to 20% of patients hospitalized in vaso-occlusive pain crisis, often caused by fat embolization syndrome or pneumonia.
Secondary pulmonary hypertension, defined by right heart catheterization and often unrecognized, occurs in 10% of adult patients with sickle cell disease.
Red cell transfusion is an important treatment for most patients with sickle cell disease requiring intensive care management.
Rapid exchange transfusion is indicated for central nervous system events, serious respiratory disease, or multiorgan failure.
Transfusion management in patients with sickle cell disease requires investigation of alloimmunization history.
Preoperative red cell transfusion and detailed supportive care are advisable for significant surgery in patients with sickle cell disease.
Sickle cell disease is a highly prevalent disease in the United States, affecting 1 in 500 African American infants. It is common in individuals of African, Caribbean, Mediterranean, Arab, and other Middle Eastern descent. It is a genetic disorder with an autosomal recessive inheritance pattern. Sickle cell disease is often called “the first molecular disease” because the biochemical alteration in sickle hemoglobin described by Linus Pauling in 1948 was one of the first lesions identified at the molecular level for a human disease. Sickle hemoglobin forms rod-like polymers in deoxygenated red cells in areas of the circulation, with low oxygen tension, acidosis, or hyperosmolarity. Sickle hemoglobin polymerization causes a host of secondary molecular and cellular changes, many of which impair blood flow and contribute to tissue damage. The microcirculation can be acutely or chronically impaired in virtually any organ in the body, resulting in the characteristic crisis pattern of intermittent pain and acute organ injury superimposed on the gradual development of chronic organ failure.
Despite the early progress in a molecular understanding of sickle cell disease, its treatment remained largely palliative for many decades. In recent years, the longevity of patients with sickle cell disease has been prolonged by the institution of prophylactic penicillin treatment and immunization to decrease mortality rate from pneumococcal sepsis. Chronic transfusion therapy for selected patients has improved outcome, and acute transfusion therapy is the central intervention for most complications requiring admission to the intensive care unit (ICU). Hydroxyurea, the first treatment approved by the Food and Drug Administration for sickle cell disease, decreases disease severity and mortality rate.1,2 Most recently, new paradigms have emerged regarding the pathophysiology of secondary complications of sickle cell disease, with much of this involving the pathological effects of intravascular hemolysis which impairs normal vascular function, in part by disrupting the nitric oxide (NO) pathway3-5 (Fig. 96-1). These new paradigms have triggered a wave of research seeking to translate these basic science advances into clinical practice.6 This chapter reviews general aspects of the genetics and pathophysiology of sickle cell disease, common clinical problems with an emphasis on those faced in a critical care setting, and contemporary therapeutic approaches to these complications of sickle cell disease.
GENERAL BACKGROUND
In 1910, James Herrick first reported the observation of sickle-shaped red cells from the blood of an anemic Chicago dental student from Grenada. Subsequently, in 1949, Linus Pauling and his colleagues demonstrated a difference in the electrophoretic pattern of sickle hemoglobin compared with normal hemoglobin and introduced the label of “molecular disease.” Marotta and colleagues in 1977 showed that the hemoglobin alteration was due to a single nucleotide change in the gene encoding the β subunit of hemoglobin A.
GENETICS AND EPIDEMIOLOGY
Sickle cell disease is a general category of sickling disorders including the most common homozygous (HbSS) form, which is called sickle cell anemia. Sickle cell anemia is inherited in a classic Mendelian autosomal recessive pattern, with affected individuals demonstrating complications of hemolytic anemia and impaired microvascular blood flow. Usually the heterozygous carrier state, sickle trait, is completely asymptomatic, although rarely vaso-occlusive events, splenic infarction, renal papillary necrosis, or sudden death can occur, usually under high-altitude hypoxic conditions. Several other mutant β-globin alleles can induce sickling disorders when inherited in combination with the sickle trait, the most common of which are β-thalassemia and hemoglobin C. Although the homozygous state typically results in severe disease, the sickle trait appears to confer resistance to severe malaria, leading to an evolutionary selection for this carrier state in geographic regions with a high prevalence of malaria.7 The sickle trait allele appears to have independently arisen at least three times in Africa and once in the Arab-Indian region. Migration from these regions has brought sickle cell disease to other parts of the world, particularly the Caribbean, Mediterranean, and the North and South American regions. Some variation in disease severity has been attributed to distinct haplotypes surrounding these alleles; additional polymorphisms in several other genes appear to modify disease severity, including those affecting expression of the α- and β-globin (thalassemia mutations) and γ-globin (fetal hemoglobin) genes.
Homozygous sickle cell disease is also referred to as SS disease or sickle cell anemia, to distinguish it from hemoglobin SC disease, a compound heterozygous combination of sickle trait and hemoglobin C trait. Hemoglobin SC disease is clinically similar in scope to SS, but hemoglobin SC disease on average presents milder or less frequent vaso-occlusive complications. The spectrum of SC disease overlaps considerably with SS. Other compound heterozygous forms of sickle cell disease are the S-β-thalassemias and S-α-thalassemias. A combination of a β-globin S allele with a nonexpressing β-globin allele, called S-β0–thalassemia, results in production of only hemoglobin S, and the clinical phenotype closely resembles SS. If the thalassemia allele of β-globin permits partial expression of a normal β-globin protein, the combination is called S-β+–thalassemia, and like hemoglobin SC, the disease tends to be milder. There are several additional combinations of the β-globin S allele with rare variant hemoglobins that can result in different severities of sickle cell disease. With S-α-thalassemias, a reduction in the amount of α-globin produced results in hemoglobin chain imbalances that reduce the concentration of total hemoglobin in the red blood cell. Because hemoglobin S polymerization is reduced at lower intraerythrocytic hemoglobin concentrations, this reduces the severity of hemolytic anemia. In general, patients with S-thalassemias and SC disease have higher steady-state hemoglobin concentrations (10-12 g/dL) than patients with homozygous Hb-SS disease (7-9 g/dL).
PATHOPHYSIOLOGY
The single nucleotide mutation in the sixth codon of the β-globin gene changes the corresponding amino acid in β-globin from valine to glutamic acid. This induces a conformational change in the hemoglobin tetramer that renders it susceptible to polymerization with deoxygenation in hypoxic, hyperosmolar, or acidic regions of the circulation. The rod-like polymer bundles distort the red cell membrane into the characteristic sickle shape. Intracellular hemoglobin S polymerization and “sickling” are associated with a complex combination of pathophysiologic changes (Table 96-1 and Fig. 96-2). It is likely that adhesion of erythrocytes and leukocytes to endothelial receptors such as vascular cellular adhesion molecule 1 (VCAM-1) combines with physical rigidity and distortion (“sickling”) of the red blood cells to occlude the microvascular circulation. Additional involved cell adhesion molecules include intercellular adhesion molecule 1, E selectin, and P selectin. The resulting tissue ischemia-reperfusion drives tissue and organ injury, generalized inflammation, and ultimately produces tissue infarction. These pathophysiologic events produce clinical and biochemical perturbations, such as fever and leukocytosis, which can mimic sepsis. Tissue ischemia and infarction produce severe pain attacks called acute pain episode or vaso-occlusive crisis (VOC) and specific end-organ pathophysiology, such as the acute chest syndrome (ACS), an acute lung injury syndrome similar to acute respiratory distress syndrome (ARDS), discussed in greater detail below.
Pathogenesis of Sickle Cell Anemia
| Mechanism | Modifying Factors |
|---|---|
| RBC sickling | |
| Hemoglobin polymerization (crystal-solution equilibrium) | Hemoglobin deoxygenation-degree and duration of hypoxia |
| Hemoglobin concentration-cellular dehydration | |
| Inversely proportional to hemoglobin F concentration | |
| Disturbance of vasoregulation | Transcriptional induction of endothelial cell genes encoding endothelin 1, a potent vasoconstrictor, after contact with sickled RBCs |
| Endothelin 1 levels are elevated during VOC and ACS | |
| Cell-free plasma hemoglobin impairs nitric oxide bioavailability | |
| Activation of the coagulation system | Platelet activation by hemolysis and cell free hemoglobin |
| Pulmonary in situ thrombosis and thromboemboli from distant sources | |
| Elevated whole blood tissue factor procoagulant activity | |
| Increased adhesion of RBCs and WBCs to vascular endothelium | Increased expression of adhesion molecules results in RBC-endothelial cell adhesion and endothelial cell damage |
| Association between clinical severity and in vitro adhesion of sickled RBCs to endothelial cells | |
| Increased expression of adhesion molecules after activation by inflammatory cytokines (TNF α and IL-1β) | |
| Reticulocyte integrin complex, α4β1, binds to plasma fibronectin and endothelial cell VCAM-1 | |
| Sickle red blood cell CD36 and endothelial cell CD36 bind to thrombospondin secreted by activated platelets | |
| Increased numbers of circulating microvascular endothelial cells in sickle cell disease, particularly during VOC | |
| Increased endothelial cell activation as evidenced by increased ICAM-1, VCAM-1, E selectin, P selectin |
FIGURE 96-2
Model of pathophysiology of sickle cell disease. This model reflects a summary of pathophysiologic events for which there is substantial supportive evidence. Nitric oxide (NO·) is normally synthesized from l-arginine by isoforms of NO synthase (NOS). Recent findings have implicated vascular instability due to inactivation of endogenous NO by free plasma hemoglobin released from lysed red cells and by superoxide (O2−) generated possibly by xanthine oxidase (XO), which is strongly expressed in the plasma of patients with sickle cell disease. NO deficiency leads to vasoconstriction and inflammation. Cell-free hemoglobin also induces the expression of endothelin 1 (ET-1), an extremely potent vasoconstrictor. The best characterized mechanism of vaso-occlusion occurs due to extensive hemoglobin S polymerization, leading to dense, poorly compliant red cells that cannot readily traverse capillaries. More recent evidence has indicated the adhesion of immature red cells and leukocytes to cell adhesion molecules (green arcs) expressed on endothelial luminal surfaces of postcapillary venules in response to NO deficiency, tumor necrosis factor α and other inflammatory cytokines, commonly found to be in patients with sickle cell disease. Activated monocytes and platelet-monocyte aggregates have been described in patients with sickle cell disease, and these appear to secrete inflammatory cytokines. The resulting sluggish flow of desaturated red cells in the venules may promote sickling, leading to multifactorial vaso-occlusion.
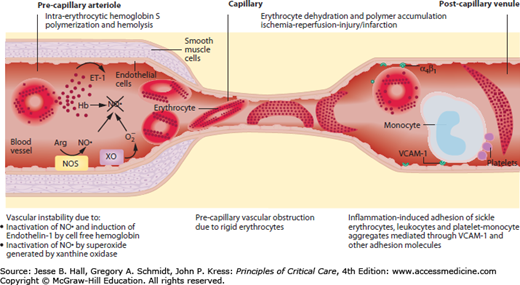
Steady-state sickle cell disease is characterized by intense inflammation that accelerates during VOC, likely secondary to nearly constant tissue ischemia-reperfusion. This is illustrated by frequent fever during VOC and the ACS and by increased leukocyte, platelet, and endothelial cell activation, with increased circulating mediators and markers of oxidant stress and inflammation. Examples of these inflammatory markers include elevated levels of C-reactive protein, cytokines (tumor necrosis factor α, interleukin 1β, and granulocyte-macrophage colony-stimulating factor), chemokines (interleukin 8), integrins (α4β1), selectins (E and P), adhesion molecules (VCAM-1 and intracellular adhesion molecule 1), markers of oxidant stress and inflammation (isoprostanes and tyrosine nitration), hemostatic activation (platelets and coagulation factors), angiogenic factors (placenta growth factor), and vasoconstrictors (endothelin 1).4,5
Associated with this inflammation is a state of endothelial dysfunction, with impaired NO bioavailability. Strong evidence points to intravascular consumption of NO by cell-free plasma hemoglobin liberated from red cells by intravascular hemolysis and by radical-radical inactivation reactions with superoxide produced by xanthine oxidase, uncoupled endothelial NO synthase and the NADPH oxidases.8,9 Impairment of NO signal transduction leads to many adverse consequences, including increased inflammation and impaired microcirculatory blood flow.10,11 It may also lead to platelet activation and acute and chronic pulmonary hypertension.12,13
BASELINE PHYSIOLOGY
Sickle cell disease is fundamentally a severe hemolytic anemia. This is characterized by a normocytic, normochromic anemia with reticulocytosis at baseline. Characteristic laboratory profiles for patients with SS and SC without hydroxyurea treatment are presented in Table 96-2. Hemolysis is mostly but not exclusively extravascular and is due primarily to mechanical injury and destruction of erythrocytes rendered rigid by intracellular hemoglobin S polymerization. Baseline leukocytosis is commonly present and should not necessarily be construed as evidence of infection. Nucleated red blood cells commonly are miscounted in the complete blood count as white cells, although the overall counting error is usually relatively small. The diminished oxygen-carrying capacity due to chronic anemia is compensated by increased cardiac output (Table 96-3). Cardiomegaly and biventricular chamber dilation due to constantly increased cardiac output are common (Table 96-4). Splenic dysfunction due to sub-clinical splenic infarction is almost universal by late childhood in patients with homozygous sickle cell disease; on computed tomography, the spleen appears typically as a small calcified mass (Fig. 96-3). As discussed below, patients with SC disease or S-thalassemia may retain their spleens and present with acute splenic sequestration or infarction. While almost 98% of adult SS patients have functional asplenia, about 50% of patients with SC or S-β+-thalassemia have a spleen. Likewise, gradual infarction of the renal medulla by adolescence leads to isosthenuria (iso-osmolar urine), with increased urination because of diminished urinary concentrating capacity, mimicking a mild nephrogenic diabetes insipidus phenotype. Aside from increasing maintenance fluid requirements, these changes mean that urine specific gravity is not closely indicative of hydration status. The increased cardiac output and higher plasma content of each milliliter of blood present the renal glomeruli with a volume of plasma per minute up to twice that of patients without sickle cell disease. This leads to baseline glomerular hyperfiltration (up to 200 mL/min) and a low baseline serum creatinine range in adults of approximately 0.5 to 0.6 mg/dL, and even lower in children (see Table 96-2). Serum creatinine levels above this level actually imply renal insufficiency. A serum creatinine level of 1.0 mg/dL may often be associated with other consequences of renal insufficiency, such as hyperkalemia, hyperuricemia, and increased anemia due to a partial defect in erythropoietin secretion. Importantly, these features of renal insufficiency may arise even when the glomerular filtration rate may be in the 80-mL/min range. However, renal physiology is more normal in SC disease or S-β+-thalassemia.
Ranges of Laboratory Values in Normal Adults With Sickle Cell Diseasea
| Parameter | SS | SC | General Population |
|---|---|---|---|
| Oxygen saturation (%) | 95.4 ± 3.3 | 98.3 ± 1.2 | 95-98 |
| Leukocyte count (K/µm3) | 11.3 ± 3.2 | 8.7 ± 4.0 | 4.5-11.0 |
| Hemoglobin (g/dL) | 8.3 ± 1.1 | 11.6 ± 1.2 | M: 13.5-17.5 |
| F: 12.0-16.0 | |||
| Platelets (K/µm3) | 401 ± 119 | 268 ± 117 | 150-450 |
| Urea nitrogen (mg/dL) | 9 ± 6 | 9 ± 5 | 6-20 |
| Creatinine (mg/dL) | 0.7 ± 0.3 | 0.8 ± 0.2 | 0.7-1.2 |
| Alkaline phosphatase (U/L) | 115 ± 91 | 95 ± 51 | 25-100 |
| Bilirubin, total (mg/dL) | 3.5 ± 1.8 | 1.7 ± 1.1 | 0.3-1.2 |
| Bilirubin, direct (mg/dL) | 0.5 ± 0.3 | 0.4 ± 0.6 | 0-0.2 |
| Lactate dehydrogenase (U/L) | 437 ± 154 | 247 ± 80 | 208-378 |
| Ferritin (ng/mL) | 656 ± 1058 | 253 ± 395 | M: 20-250 |
| F: 10-120 | |||
| Iron (µg/dL) | 94 ± 41 | 87 ± 57 | M: 65-175 |
| F: 50-170 | |||
| Transferrin (mg/dL) | 215 ± 57 | 249 ± 46 | 200-400 |
Echocardiographic Measurements in Adults With Sickle Cell Disease With or Without Pulmonary Hypertensiona
| Parameter | Without Pulmonary HTN | With Pulmonary HTN | Normal Range |
|---|---|---|---|
| LA size (mm)b | 39 ± 5 | 45 ± 8 | <40 |
| LV size (mm)b | 37 ± 7 | 40 ± 7 | 37-56 |
| RA area (cm2)b | 17 ± 4 | 20 ± 5 | <18 |
| Ejection fraction (%) | 61 ± 5 | 60 ± 9 | 55-74 |
| Systolic blood pressure (mm Hg)b | 119 ± 15 | 129 ± 21 | 90-135 |
| Diastolic blood pressure (mm Hg) | 67 ± 11 | 68 ± 13 | 50-90 |
| Mean arterial pressure (mm Hg)c | 85 ± 11 | 88 ± 15 | 70-105 |
| Tricuspid regurgitant jet velocity (m/s) | 2.0 ± 0.4 | 2.8 ± 0.3 | <2.5 |
| Systolic pulmonary artery pressure, estimated (mm Hg) | 26 ± 6 | 42 ± 8 | 19-37 |
Cardiac Catheterization Data for Adults With Sickle Cell Diseasea
| Parameter | Without Pulmonary HTN | With Pulmonary HTN |
|---|---|---|
| Pulmonary artery systolic (mm Hg) | 30.3 ± 5.77 | 54.3 ± 12.26 |
| Pulmonary artery diastolic (mm Hg) | 11.7 ± 4.86 | 25.2 ± 7.72 |
| Pulmonary artery mean (mm Hg) | 17.8 ± 4.86 | 36.0 ± 7.78 |
| Cardiac output (L/min) | 8.62 ± 3.03 | 8.60 ± 1.76 |
| Pulmonary capillary wedge pressure (mm Hg) | 10.6 ± 3.82 | 16.0 ± 5.87 |
| Pulmonary vascular resistance (dynes·s/cm5) | 57 | 162 |
FIGURE 96-3
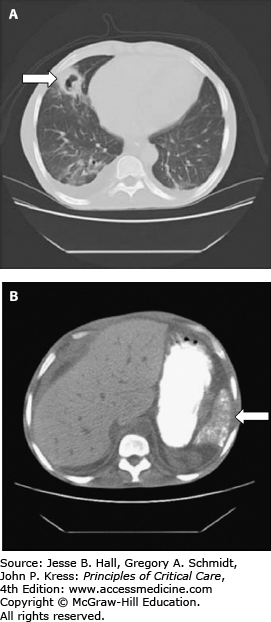
Sickle cell disease injury to soft tissues. A. Lung cavitation resulting from previous acute chest syndrome, causing infarction and necrosis of a subsegmental region of the left middle lobe. The cavitary lesion (arrow) has an inflammatory wall that subsequently resolved after antimicrobial therapy. B. The calcified, atrophic spleen (arrow) is visible on an abdominal scan from the same patient. This is typical in adults with homozygous sickle cell disease. The stomach is visualized by oral contrast material.
These baseline physiologic perturbations result in a hyperdynamic cardiovascular state with relatively low blood pressure, low systemic and pulmonary vascular resistances, high cardiac output and renal hyperfiltration (see Tables 96-3 and 96-4). These findings resemble other high cardiac output states such as pregnancy and thyrotoxicosis. These hemodynamic parameters combined with significant baseline inflammation in patients with sickle cell disease, characterized by leukocytosis and increased serum ferritin, often can be mistaken for sepsis in the absence of a true infectious insult (see Table 96-4).
COMMON CLINICAL PROBLEMS
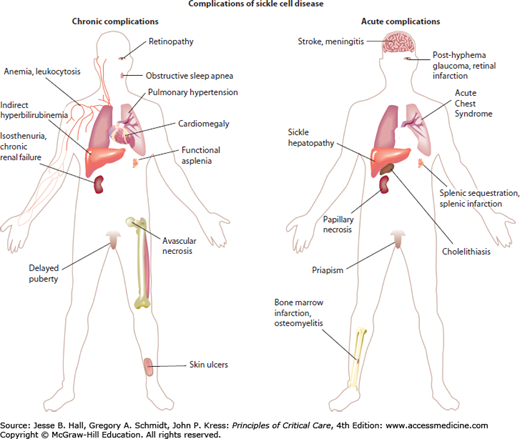
Common and significant clinical problems in patients with sickle cell disease are summarized in Figure 96-1. Virtually every organ system is subject to acute ischemia-reperfusion injury and infarction during vaso-occlusive events. Many organ systems are subject to chronic dysfunction, particularly in the adult aging sickle cell disease population.
VOC is perhaps the most characteristic manifestation of sickle cell disease. These severe painful attacks, most frequently affecting the extremities and back, result from acutely impaired microvascular blood flow. Most commonly affected are bones, due primarily to ischemia and infarction of the bone marrow and inflammation of the periosteum, with intense bone pain due to the resulting edema and tissue swelling in the inelastic compartment of the bone marrow medulla. Most often, pain affects the lumbosacral vertebrae, iliac and the long bones, although any bone may be affected. Joint effusions of the elbows and knees are common. At times, especially in children, there is difficulty in distinguishing painful crisis from osteomyelitis, another complication of sickle cell disease. The mainstays of treatment for pain crisis are analgesics, in particular nonsteroidal anti-inflammatory drugs and opioids. Correction of dehydration or hypoxemia is important, as is careful monitoring for development of acute chest syndrome (ACS; see below). Clinicians frequently undertreat the severe pain of sickle cell crisis. Opioid tolerance due to recurrent or chronic opioid treatment may dramatically increase the dosage required in individual patients. There is little, if any, risk for developing opioid dependency with short courses of large-dose opioids for VOC.
Ischemic stroke is a frequent complication of sickle cell disease, occurring at any age, but mainly in children. This topic is discussed in detail below in the section on special problems in the ICU.
A proliferative retinopathy develops in 10% of patients with SS disease and at least twice as often in patients with SC disease. This chronic complication may be treated with laser photocoagulation. Two acute complications present ophthalmologic emergencies. The first is vision loss due to acute occlusion of the central retinal artery. This may resolve with emergency exchange transfusion before retinal infarction occurs. The second emergency is traumatic hyphema in a patient with sickle cell disease or sickle trait. The environment of the anterior chamber of the eye promotes sickling, and this can readily cause acute glaucoma. Evacuation of the blood should be undertaken for hyphema without delay to avoid vision loss.14
Sickle cell disease is associated with a high incidence of acute and chronic lung complications. The ACS is an acute lung injury syndrome and, in cases of extensive lung involvement, is similar to ARDS. It is defined by a new pulmonary infiltrate secondary to alveolar consolidation, not atelectasis, in a patient with sickle cell disease often accompanied by chest pain, fever, tachypnea, wheezing, or cough. It is caused frequently by pulmonary fat embolism syndrome from infarcted, necrotic bone marrow or by an exuberant pulmonary inflammatory response to common respiratory pathogens. Its pathogenesis and treatment are discussed in detail in the section on special ICU problems. Chronic complications can include the development of restrictive lung disease, pulmonary fibrosis, or pulmonary hypertension; with the latter occurring in approximately 10% of adults with sickle cell disease.15 Pulmonary hypertension appears to be the most serious risk factor for early mortality in adult patients with sickle cell disease and, in the past, may have been underrecognized by clinicians. Reactive airway disease or asthma occurs with increased frequency in children with sickle cell disease.16 Both pulmonary hypertension with right heart failure and reactive airways disease commonly complicate the acute chest syndrome and lead to a more severe clinical course.16
Although vaso-occlusion of the coronary microcirculation can produce myocardial infarction, acute wall motion abnormalities, and cardiac enzyme release, coronary atherosclerosis is essentially nonexistent in patients with sickle cell disease for reasons that are poorly understood, possibly owing to low levels of low-density lipoprotein cholesterol, early mortality of males, and increased heme oxygenase-1 activity. Cardiomegaly and biventricular chamber dilation are nearly universal due to a long-term high cardiac output state as compensation for severe chronic anemia. High-output cardiac failure, diastolic dysfunction, and pulmonary hypertension are common cardiac complications of sickle cell disease (see Tables 96-3 and 96-4).
Up to 42% of patients with sickle cell disease have calcium bilirubinate gallstones by age 18 years, although only a fraction of these patients are symptomatic. Stones develop due to hemolytic anemia and the very high rate of bilirubin excretion in bile as a breakdown product of hemoglobin turnover. In cases of acute cholecystitis, awaiting its resolution decreases perioperative complications. Acute vaso-occlusion in the liver can cause pain, elevated transaminases, and extreme hyperbilirubinemia, generally responding well to supportive care. Rarely, the liver may acutely sequester peripheral blood cells, clinically analogous to splenic sequestration syndrome (see Special Problems in the ICU).
Nearly all patients with sickle cell disease develop progressive loss of splenic function secondary to its microvascular vaso-occlusion and infarction, beginning at birth and complete by age 10 years. This manifests primarily as a marked susceptibility to sepsis and meningitis due to encapsulated organisms, particularly in children younger than 5 years. The incidence of serious infection is dramatically decreased by penicillin prophylaxis and conjugate vaccines against Haemophilus influenzae and Streptococcus pneumoniae. However, splenic dysfunction still causes a modest lifelong risk of overwhelming sepsis. The spleen is atrophic and nonfunctional in 98% of adults with homozygous SS sickle cell anemia (see calcified spleen in Fig. 96-3B). A dramatic acute complication of sickle cell disease is splenic sequestration crisis, described in detail in the section on special problems in the ICU. This complication occurs frequently in pediatric patients with functioning spleens, primarily young children with homozygous SS disease, and adults with hemoglobin SC sickle cell disease or S-β+-thalassemia because approximately 50% of these patients retain their spleen into adulthood. It is conceivable that hydroxyurea therapy in children with sickle cell disease might lead to prolongation of splenic function.
Isosthenuria or hyposthenuria (decreased urine osmolarity) develops in most patients by age 10 years, resulting in increased maintenance fluid and sodium requirements. Hematuria due to papillary necrosis is an occasional complication, usually self-resolving. A nephropathy with nephrotic grade proteinuria can gradually progress to uremia. Early signs include the normally low serum creatinine exceeding 0.6 mg/dL, progressively severe anemia, and a rise in the serum uric acid level. Angiotensin-converting enzyme inhibitors can decrease proteinuria and potentially slow progression of renal insufficiency. Uremia has been treated with dialysis and with renal transplantation.
Bone marrow infarcts are frequent causes of pain. These may be detected on magnetic resonance imaging or radionuclide imaging with technetium sulfur colloid, although it is not normally clinically helpful to ascertain these by imaging. The heads of the femur and humerus are susceptible to avascular necrosis, a potential source of constant pain and disability, sometimes requiring joint replacement. Ischemic bone becomes susceptible to bacterial osteomyelitis; however, this is quite rare in adult patients. Staphylococcus aureus is the most common organism in sickle cell osteomyelitis episodes. Salmonella
Related posts:






Full access? Get Clinical Tree


