KEY POINTS
Acute-on-chronic respiratory failure (ACRF) occurs when often minor, although commonly multiple, insults cause acute deterioration in a patient with chronic respiratory insufficiency.
ACRF is usually seen in patients known to have severe chronic obstructive pulmonary disease (COPD), but occasionally it manifests as cryptic respiratory failure or postoperative ventilator dependence in a patient with no known lung disease.
The wide variety of causes of ACRF may be compartmentalized into causes of incremental load, diminished neuromuscular competence, or depressed drive, superimposed on a limited ventilatory reserve.
Intrinsic positive end-expiratory pressure (PEEPi) is a central contributor to the excess work of breathing in patients with ACRF.
The most important therapeutic interventions are administration of oxygen, bronchodilators, corticosteroids, and noninvasive positive-pressure ventilation (NIV).
NIV can be used in most patients to avoid intubation and has been shown to improve survival.
The decision to intubate a patient with ACRF benefits from clinical judgment and a bedside presence. Hypotension and severe alkalemia commonly complicate the immediate periintubation course, but they are usually avoidable. However, delaying intubation when NIV is ineffective may worsen outcomes.
Ventilator settings should mimic the patient’s breathing pattern, with a modest respiratory rate (eg, 20/min) and small tidal volume (eg, 450 mL); some positive end-expiratory pressure (eg, 5 cm H2O) should be added.
Prevention of complications such as gastrointestinal hemorrhage, venous thrombosis, and nosocomial infection is a crucial component of the care plan.
The key to liberating the patient from the ventilator is to increase neuromuscular competence while reducing respiratory system load.
In selected patients, extubation to NIV despite failed spontaneous breathing trials reduces ventilator and ICU days and further improves survival.
In the past three decades, mortality from chronic obstructive pulmonary disease (COPD) has risen dramatically,1 making chronic lower respiratory disorders the third leading cause of death in the USA in 2009.2 COPD was fifth internationally in 2002 and projected to be the fourth leading cause of mortality by 2030.3 Compared with people with normal lung function, subjects with severe COPD (FEV1 <50% predicted) followed for 22 years as part of the National Health and Nutrition Examination Survey (NHANES I) had a 2.7-fold increased risk of death (95% confidence interval [CI] 2.1-3.5) in an adjusted analysis.4 This trend is apparent in men and women, more prominent in black Americans, and clearly related to cigarette smoking. More women than men have died of COPD in the USA since 2000.4,5 Internationally COPD bears a significant morbidity and mortality burden accounting for 27,700 disability adjusted life years (DALYs).6 Admissions to ICUs for exacerbations of COPD account for a substantial portion of bed-days,7 since these patients often require prolonged ventilatory support. Between 1998 and 2008 in the USA, there were an average of 765,067 (95% CI 764,360-765,773) hospitalizations for acute exacerbation of COPD of which 8.1% required a period of respiratory support8; 13.2% of patients with respiratory failure requiring mechanical ventilation in a recent survey USA survey of 180,326 hospitalizations had significant comorbid pulmonary disease.9 In surgical ICUs, COPD is an important problem as well, since it is one of the more common reasons for a prolonged postoperative recovery. An approach to this disease is an essential component of the intensivist’s armamentarium.
This chapter describes the pathophysiology and management of patients with chronic pulmonary disease (most with COPD) who require intensive care for decompensation of their normally precariously balanced ventilatory state. This acute deterioration superimposed on stable disease is termed acute-on-chronic respiratory failure (ACRF). Patients may present to the ICU with worsening dyspnea, deteriorating mental status, or respiratory arrest. Especially when there is a preexisting diagnosis of lung disease, the diagnosis of ACRF can be made easily. However, it is important to remember that not all patients with severe COPD will have been so identified. In many patients with respiratory distress, congestive heart failure or pulmonary thromboembolism is considered first; making a correct diagnosis of ACRF requires a high index of suspicion. On occasion, the disease is even more occult, for example, in a postoperative patient who fails extubation and then is noted to have hyperinflation on the chest radiograph. Since optimal therapy depends on accurate diagnosis, underlying COPD should be part of the differential diagnosis for most patients with dyspnea or inability to sustain unassisted ventilation.
A severe acute exacerbation of COPD (AECOPD) is characterized by a sustained worsening from the stable state that is acute in onset and requires hospitalization.10,11 The typical symptoms are dyspnea that has been worsening over days, often with increased cough and sputum production. Physical examination typically demonstrates respiratory distress, accessory muscle use, a prolonged expiratory time, recruitment of expiratory muscles, and wheezing. As discussed below, the absence of respiratory distress is not necessarily reassuring and when associated with somnolence is a grave and ominous sign of impending respiratory arrest. The chest radiograph is usually abnormal, reflecting the chronic lung disease, but only in 15% to 20% of cases reveals an acute finding (eg, pneumonia, pneumothorax, pulmonary infarction, pulmonary edema) that results in a change of management.12 Sometimes there are indicators of acute infection, such as purulent sputum, fever, leukocytosis, and a new radiographic infiltrate. Typical initial arterial blood gas values on room air show a PO2 of 35 to 45 mm Hg and a PaCO2 of 60 to 70 mm Hg. Comparison with values obtained when the patient is stable can be useful as many patients have compensated metabolic acidosis with chronically elevated PaCO2 at baseline. Electrocardiography (ECG) may show signs of right atrial enlargement or right ventricular hypertrophy and strain. P-wave amplitude >1.5 mm is universal in patients with AECOPD (but not necessarily ACRF), although classical P pulmonale (P-wave amplitude in leads II, III, and/or aVF >2.5 mm) is uncommon. Resolution of the exacerbation is associated with an amplitude reduction of approximately 0.8 mm.13 Thus serial ECG may be useful in assessing response to therapy.
Although the short-term risk of death is high for ACRF,8 the prognosis for patients with ACRF is not uniformly poor, despite severe underlying pulmonary impairment. In a prospective analysis of 250 admissions (180 patients) to an ICU for acute respiratory failure complicating COPD, hospital mortality was 21% and was strongly associated with the development of extrapulmonary organ failures.14 However, more recent analyses of patients discharged after an episode of ACRF complicating AECOPD in the USA between 1998 and 2008 revealed a bimodal mortality rate. Chandra and colleagues reported that while mortality for patients requiring intubation and MV (mechanical ventilation) remained at 22%, the rate for patients managed exclusively with NIV was closer to 6% in 2008, an approximately 50% reduction over the course of a decade.8 Six-month and 1-year survival following ACRF approximates 40% and 45%, respectively.15,16 These rates may in part be explained by the uniform management of ACRF patients in ICUs in North America. This is not the case in other regions. For example, a recent Scottish single center experience reported that among 275 patients treated with NIV (mean baseline pH of 7.24 and PCO2 77 mm Hg) hospital mortality was 32% and only 5% of the patients received ICU level care. All-cause 1-year mortality was 55%.17
Some patients will return to an acceptable quality of life (QOL), and some even go back to work. In a prospective cohort of 611 ambulatory COPD patients, Esteban et al18 reported that when controlled for COPD disease severity and baseline QOL, number of hospitalizations for AECOPD was an independent predictor of reduced QOL in COPD; 7% of patients required three or more admissions and experienced particularly marked deterioration in QOL over 5 years of follow-up. In a cohort of 1016 patients admitted with a COPD exacerbation and a PaCO2 >50 mm Hg, 1-year survival was 47%, but only 26% of the patients rated their QOL as good or better when surveyed at 6 months.7
Predictors of poor survival include the underlying cause of chronic respiratory failure and a high BODE index19 (an integrated assessment of body mass index, airflow obstruction, dyspnea, and exercise capacity), older age,7,15,20 more than three acute exacerbations in 5 years,20 history of congestive heart failure, cor pulmonale,7 presence of serious comorbid disease,21 lower PO2 : FiO2 ratio,7 lower serum albumin level,7,15,22 chronically elevated PCO220 development of extrapulmonary organ failures,14,15 and requirement for >72 hours of ventilation.21 However, these indicators are not sufficiently refined to allow accurate prognostication in an individual patient.
As a result, critical care resource utilization and costs are substantial. Ely and coworkers calculated that respiratory care costs were almost twice as much for patients with COPD compared with non-COPD related respiratory failure ($2422 [$1157-$6100] vs $1580 [$738-$3322], respectively; p = 0.01, $1996), despite similar ICU lengths of stay and mechanical ventilation days.23 However, attributable health care costs and resource utilization for ACRF management are influenced strongly by prevailing care models. In 2008, hospital charges for patients managed for ACRF complicating AECOPD were approximately $35,000 if care with NIV alone was successful. However, costs increased to more than $100,000 if invasive MV was required during the hospitalization.8 Significant efficiencies have been reported when noninvasive ventilation is administered in ward-based settings24 although outcomes may be less satisfactory.17
Ideally, patients followed in the clinic with known, severe COPD will be encouraged to discuss with their physicians their wishes regarding intensive care before acute deterioration. Unfortunately, this is only occasionally accomplished.25 It is our approach to fully support patients with ACRF who believe their QOL is acceptable and have an appreciation of the burden and potential outcomes of ICU treatment,26 especially since most will be managed successfully with noninvasive ventilation, and most of those intubated will eventually be successfully liberated from the ventilator and survive to hospital discharge.15,16,27 On the other hand, when mechanical ventilation seems excessive to the patient or physician, defining the goals of care as the provision of comfort and relief from dyspnea and pain is appropriate.28 We urge clinicians caring for COPD patients with compensated respiratory failure to address advance directives and desire for life-sustaining therapies during routine ambulatory clinic appointments when informed and deliberate decision making can be shared by the patient and their loved ones.
PATHOPHYSIOLOGY
Alveolar ventilation is maintained by the central nervous system, which acts through nerves and the respiratory muscles to drive the respiratory pump. The three subsets of ventilatory failure are loss of adequate drive, impaired neuromuscular competence, and excessive respiratory load. This concept is developed in Figure 54-1. The central nervous system drives the inspiratory muscles via the spinal cord and phrenic and intercostal nerves. Inspiratory muscle contraction lowers pleural pressure, thereby inflating the lungs. The pressure generated by the inspiratory muscles (neuromuscular competence) must be sufficient to overcome the elastance of the lungs and chest wall and abdomen (elastic load), as well as the flow resistance of the airways (resistive load). Spontaneous ventilation can be sustained only as long as the inspiratory muscles are able to maintain adequate pressure generation.29
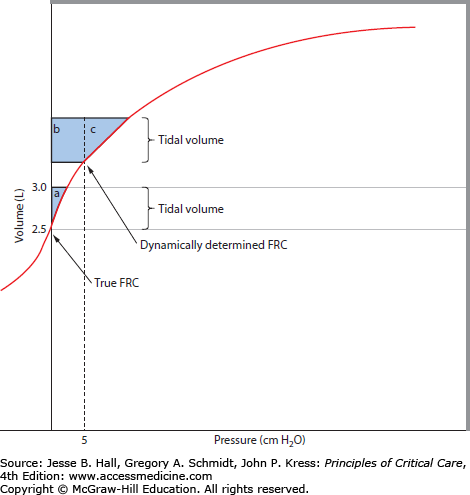
FIGURE 54-1
The effects of PEEPi on work of breathing. A volume-pressure curve for the respiratory system is shown. Under normal conditions, alveolar pressure is atmospheric at end expiration (shown as true FRC). As transpulmonary pressure is generated by the respiratory muscles, there is a VT change (here, from 2.5 to 3.0 L, a VT of 0.5 L). The pressure-volume product or the external work is shown by shaded area a. With gas trapping due to airflow obstruction, FRC can become dynamically determined. In this example, the end-expiratory pressure (PEEPi) is 5 cm H2O, which raises the end-expired lung volume. Now, for the same VT generation, the respiratory muscles must overcome the positive alveolar pressure (PEEPi) before flow occurs, and the work of breathing is increased accordingly (b + c). In addition, the VT change can occur on a flatter portion of the volume-pressure curve, resulting in yet another increment in elastic load. The addition of continuous positive airway pressure (CPAP) in an amount to counterbalance the PEEPi has the ability to reduce the work of breathing from b + c to c only.
Relatively few patients develop ventilatory failure from loss of drive. Most often this occurs in the setting of drug or alcohol overdose or physician-directed sedation. Typically these patients present little challenge, requiring only supportive care until the drug can be reversed or metabolized. One exception is the group of patients with undiagnosed sleep disordered breathing. Although sleep disordered breathing is no more common in people with mild COPD than in healthy controls,30 many patients with chronic respiratory failure have a significant component of central apnea often in conjunction with left ventricular or biventricular cardiac dysfunction. This presents with nocturnal desaturation. Specific therapy directed at intensive heart failure management and relief of their sleep disorder, including nocturnal bilevel pressure support,31 adaptive servoventilation32 (a form of closed-loop pressure targeted ventilation used for central sleep apnea that provides breath-by-breath adjustment of inspiratory pressure support), and supplemental oxygen, may be required. Far more common and difficult are patients who have adequate drive but have inadequate neuromuscular function, excessive load, or both.
In the typical AECOPD patient who is dyspneic, tachypneic, diaphoretic, and using accessory muscles of respiration, impairment of drive is clearly not the cause of ACRF. When drive has been assessed in this setting, it is greatly elevated,33 as it is in most patients who fail to be “weaned” from mechanical ventilation.34 Nevertheless, there are patients in whom new CNS insults or drug effects contribute to respiratory failure. Even small doses of sedatives or narcotics may cause respiratory failure when superimposed on chronic ventilatory insufficiency; a careful history is essential to exclude this possibility. Occult hypothyroidism is not rare in the elderly, particularly in women. Thyroid function testing should be part of the routine screening of patients with ventilatory failure attributed to decreased drive.
It has been proposed that central depression of drive contributes to the terminal stages of respiratory failure, irrespective of the precipitating factor. According to this “central wisdom” hypothesis, overworked respiratory muscles reach a threshold of loading at which point muscle injury results in the elaboration of inhibitory signals, which feed back to the CNS to reduce drive, thereby protecting the muscles from fatigue.35-37 The relevance of this mechanism to respiratory failure remains to be demonstrated.
Central to understanding the pathophysiology of ACRF in COPD is that respiratory muscles develop impaired force generation. This can arise as a result of shortening (through classical length tension relationships) or fatigue. In health, neuromuscular function far exceeds that necessary to sustain ventilation against the normally small load. Dramatic increments in load (as in status asthmaticus) or decrements in strength (as in the Guillain-Barré syndrome) are required to cause hypoventilation. In patients with COPD, however, the respiratory system load, as judged by the ˙VO2resp, is elevated to 17% to 46% of the total body ˙VO2,38 owing to abnormal airway resistance from bronchospasm, airway inflammation, and physical obstruction by mucus and scarring and increased lung elastance from dynamic hyperinflation.39
Hyperinflation is particularly disadvantageous since it disproportionately reduces the capacity of the muscles to produce negative inspiratory pressure40 and if sufficiently severe may result in neuromechanical dissociation so that the patient demonstrates high neural drive, experiences dyspnea but does not generate additional negative (inspiratory) pressure. Patients receiving mechanical ventilation for ACRF in COPD typically have acute-on-chronic hyperinflation due to intrinsic PEEP41 compounding their pre-intubation lung function impairment. This hyperexpansion forces the inspiratory muscles to operate in a disadvantageous portion of their force-length relationship.
The role of respiratory muscle fatigue in the pathogenesis of ACRF is complex. Muscle fatigue is the reversible loss of force generation despite adequate neural stimulation. Fatigue is itself a cause of muscle weakness and can be short lasting (high-frequency fatigue), or long lasting (low-frequency fatigue), which can persist for days to weeks. In healthy adults, experimental induction of low-frequency respiratory muscle fatigue does not impair maximum ventilatory42 or exercise43 performance.
However, in ACRF low-frequency fatigue may have significant functional consequences. To the extent that fatigue is central to the development of respiratory failure, it may be caused by an inadequate supply of nutrients, excess generation of metabolites such as lactate or hydrogen ion, or depletion of muscle glycogen. Evidence to support the importance of blood supply in the genesis of fatigue comes from studies of respiratory failure in animals with hemorrhagic, cardiogenic, or septic shock.44,45 In these animals, fatigue is hastened by circulatory insufficiency. The magnitude of blood flow to the respiratory muscles seems to be important beyond aerobic needs, as demonstrated in experiments in which flow was manipulated independently of oxygen delivery.46 Hypoperfusion states, hypoxemia, and severe anemia have the potential to contribute to muscle fatigue and thereby to hasten respiratory failure.
Notably, moderately severe COPD patients do not develop low-frequency diaphragmatic fatigue after maximal exercise effort when measured with nonvolitional magnetic coil–induced twitches.47,48 Consistent with this is that low-frequency diaphragm fatigue has not been demonstrated in patients receiving mechanical ventilation for ACRF in AECOPD.49 This is not surprising given that muscle shortening protects against fatigue both in isolated models50 as well as in vivo in COPD.51 In addition intracellular modifications of cell type, 52 contractile proteins such as titin,53 and single-fiber contractile energetics54 would predict that, compared to other muscles, the diaphragm would be fatigue resistant. These cellular changes are discussed below.
By contrast, accessory muscles of respiration that are frequently recruited for expiration during AECOPD, including abdominal wall muscles, are fatigable in COPD patients during loaded exercise.55 Several experiments in humans indicate that the work intensity required of the contracting respiratory muscles, as well as their strength, are crucial factors determining fatigue.56 Additionally myocyte ischemia can result from hypotension, reduced cardiac output or “steal” of blood flow by other organs. Thus prolonged pathological loading from superimposed infection, heart failure, or other precipitants could result in sarcomeric disruption,57 cellular acidosis, and accessory (expiratory) muscle fatigue58 in the face of increased respiratory system resistance.59
Through whatever combination of causes, once respiratory muscle force generation fails, neuromuscular competence is unable to sustain the mechanical load imposed on the respiratory system. The consequence is a rapid-shallow breathing pattern39 and ultimately failure of alveolar ventilation ensues. The intensivist must therefore address both the precipitant and resultant respiratory muscle failure for treatment to be successful.
Airflow obstruction, compounded by decreased elastic recoil in patients with emphysema, leads to prolongation of expiration. When the rate of alveolar emptying is slowed, expiration cannot be completed before the ensuing inspiration. Rather than reaching the normal static equilibrium of lung and chest wall recoil at functional residual capacity (FRC) at the end of each breath, the respiratory system empties incompletely. Expiration terminates at this higher, dynamically determined FRC. At end expiration, there remains a positive elastic recoil pressure, which is called intrinsic positive end-expiratory pressure (PEEPi). Accordingly, alveolar pressure remains positive with respect to end-expiratory pressure at the airway opening, such that a greater effort must be generated by the inspiratory muscles on the subsequent breath. Intrinsic PEEP of 5 to 10 cm H2O is present in most, if not all, COPD patients with acute ventilatory failure,60-62 and PEEPi can be measured in many ambulatory outpatients as well.63 This adds a threshold load to spontaneous inspiration and, in mechanically ventilated patients, makes triggering of assisted breaths more difficult.64 Significant additional inspiratory muscle activation is required to reduce pleural pressures below PEEPi before pressure is reduced in the central airway to trigger a mechanical breath. Determinants of the magnitude of PEEPi include the degree of expiratory obstruction (including both patient and ventilator), elastic recoil, minute ventilation, and expiratory time (therefore, respiratory rate, inspiratory flow rate, and inspiratory flow profile). As discussed more fully below, counterbalancing this PEEPi with external PEEP provides a means by which to lower the work of breathing (or the work of triggering). The impact of PEEPi on the work of breathing is illustrated in Figure 54-1.
Diaphragm strength (measured by sniff esophageal pressure) in patients with severe but stable COPD is only two-thirds that of normal individuals, virtually all of this ascribable to the diaphragm position rather than to inherent muscle weakness.65 Still, patients presenting with ACRF may have not only worsening hyperinflation but also other conditions (eg, protein-calorie malnutrition,66 steroid myopathy67) that cause intrinsic muscle weakness. Even when patients with severe COPD are in a state of compensation, the increased load and diminished neuromuscular competence are precariously balanced. Only minor additional decrements in strength or increments in load are sufficient to precipitate inspiratory muscle fatigue and respiratory failure. It is this incremental deterioration in the balance of neuromuscular competence and respiratory system load that defines ACRF. Its many potential contributors are enumerated in Figure 54-2.

ADDITIONAL CAUSES OF DECREASED NEUROMUSCULAR COMPETENCE (SEE Fig. 54-2)
In order to effect adequate ventilation, the CNS must transmit drive to the working muscles via the spinal cord and peripheral nerves. Therefore, causes of neuromuscular failure, such as spinal cord lesions, primary neurologic diseases, and neuromuscular blocking drugs, may produce ACRF. Aminoglycosides68 and procainamide69 act as mild neuromuscular blockers, a feature unimportant in the great majority of patients but relevant in those with neuromuscular diseases, such as myasthenia gravis. The clinical setting may be a clue to one of the unusual causes, such as phrenic nerve injury following cardiopulmonary bypass. This occult lesion may be induced by direct trauma to the nerve or, more indirectly, by cold cardioplegia.
The most important causes of decreased neuromuscular competence and reduced force generation fall into the category of muscle weakness. In patients with COPD, respiratory muscle changes represent a balance between factors capable of impairing respiratory muscle function and metabolic adaptation of the diaphragm (Table 54-1).
Molecular and Structural Pathophysiology of Respiratory Muscle Impairment in COPD Patients With ACRF
| Respiratory Muscle Change | Impairment |
|---|---|
| Thoracoabdominal geometric changes | Changes in chest wall geometry and diaphragm position resulting from hyperinflation |
| Respiratory muscle sarcomeric changes | Deleterious shortening of diaphragm sarcomere length disturbs intrinsic sarcomeric length-tension relationship and impairs maximal force generation |
| Atrophy of thick myosin filaments | Disrupted actin: myosin ratios with increase in fast fibers (Type II myosin) |
| Disorganization of contractile myofibrils (Z-band streaming and filament misalignment) | |
| Inflammatory changes | Deleterious effects of infection and NF-κB dependent inflammation (eg, TNF-α) on skeletal muscle and systemic organ function |
| Cytosolic protein changes | Enhanced respiratory muscle proteolysis by activation of proteases (eg, calpain) via ubiquitin-dependent proteosomal degradation |
| Suppressed IGF-1/AKT-dependent protein synthesis | |
| Oxidant/antioxidant mediator changes | Reactive oxygen species generation from respiratory muscles and inflammatory cells overwhelm endogenous antioxidants (eg, catalase, superoxide dismutase) resulting in respiratory muscle injury |
| Neurologic changes | Inflammatory neuropathy |
| Reduced neuromuscular junction excitability | |
| Nutritional, pharmacological, and aging bioenergetic changes | Malnutrition, steroids, oxidant stress, insulin resistance, dysregulated sarcoplasmic calcium regulation, and aging result in reduced Type II muscle fibers (atrophy), depletion of glycogen, and high-energy phosphate and ketone accumulation and mitochondrial dysfunction leading to bioenergetic failure and impaired respiratory muscle force generation |
Changes in chest wall geometry and diaphragm position are particularly important and adversely affect the muscles of inspiration (diaphragm and intercostal muscles) and expiration (abdominal muscles). The inspiratory muscles are poorly able to tolerate maximal loading that occurs in emphysema. Hyperexpansion forces them to operate on a disadvantageous portion of their force-length curve. The piston-like displacement of the diaphragm is compromised and expansion of the lower thoracic cage is disturbed. In addition, a flattened diaphragm generates less transmural pressure for a given tension than the normally curved one, as described above. Electrolyte disturbances such as hypokalemia, hypophosphatemia,70 and hypomagnesemia71 may potentiate muscle weakness. Disturbances of the myofibrillar contractile unit have been demonstrated in humans54,72 and animal models of loaded resistive breathing that simulate ACRF. Disturbances of the sarcomere length/tension relationship,54 activation of proteolytic enzymes such as calpain that can degrade actin and intercellular adhesions,73 oxidative stress,74 and elaboration of proinflammatory cytokines have also been proposed to play a role.75
Hypophosphatemia may be exacerbated by many of the drugs used to treat ventilatory failure, such as methylxanthines, β-adrenergic agonists, corticosteroids, and diuretics, accounting for its striking prevalence in COPD patients (about 20%).76 Malnutrition, present in 40% to 50% of hospitalized patients with emphysema,77 has been suggested to be associated with respiratory muscle weakness,78 yet one study demonstrated that diaphragmatic strength was comparable between stable COPD patients with reduced body mass index (BMI 17.3 kg/m2) and those with normal BMI.79 Short-term refeeding can improve indices of respiratory muscle function66 and immune response.
Myopathy due to prolonged corticosteroid administration may contribute.67,80 Rarely, other myopathies, such as an adult variant of acid maltase deficiency or mitochondrial myopathy, may cause cryptic respiratory failure due to muscle weakness. Cellular and molecular adaptations of the respiratory musculature differ between the diaphragm and the intercostal muscles. The diaphragmatic myocytes adopt an “aerobic” fatigue-resistant Type I phenotype with prolonged loading (Table 54-1). By contrast the intercostal muscles undergo relatively greater glycolytic adaptation to chronic loading.81
Although accessory muscle fatigue is likely to contribute to the precipitation of an ACRF event, systematic evaluation has failed to demonstrate that low-frequency fatigue contributes to prolonged mechanical ventilation and failure to liberate. For example, none of 17 “weaning-failure” patients developed objective fatigue (transdiaphragmatic pressure changes with phrenic nerve stimulation), despite three-quarters of patients having a diaphragmatic tension-time index (TTdi) greater than 0.15—a level associated with diaphragmatic fatigue by several other investigators.49
ADDITIONAL CAUSES OF INCREASED LOAD (SEE FIG. 54-2)
In patients with COPD, the respiratory system load is chronically elevated owing to abnormal airway resistance and increased elastance. The increase in airway resistance is caused by bronchospasm, airway inflammation, and physical obstruction by mucus and scarring. One common cause of increased flow resistance, and the one most amenable to pharmacologic intervention, is bronchospasm. The disease course in most patients with COPD includes an asthmatic component, and bronchial hyperreactivity to provocation may be as common in COPD as in asthma.82 Exacerbations of bronchospasm increase resistive workload, precipitating ACRF. Superimposed heart failure may also cause increased airway resistance, mimicking asthma (“cardiac asthma”). Upper airway obstruction is much less common, but since these patients frequently have a history of prior intubation, tracheal stenosis should be considered. Finally, sleep disordered breathing, which commonly coexists with COPD, may need to be excluded. Especially once the patient is intubated, clues to this underlying cause of dynamic upper airway obstruction (eg, snoring) may be impossible to discern. In the proper setting (an obese, hypersomnolent patient), this possibility should be vigorously pursued. The endotracheal tube (ETT) itself presents a significant resistive workload, especially when a small size (<7.5 mm internal diameter) is chosen.83 The angulation and length of the tube and the presence of secretions also affect flow resistance. Additionally, ventilator circuit heat and moisture exchangers (“HME filters”) impose significantly greater dead space and tube resistance than heated humidifiers in ventilated ACRF patients.84 Automatic tube compensation (ATC) algorithms available on some modern ventilators are designed to maintain airway pressure by continuously calculating pressure drop across the ETT during inspiration and to decrease airway pressure during82 expiration to maintain constant alveolar pressure. However, ATC may be insufficient to compensate fully for the imposed resistive load of the ETT.85
Contributors to lung stiffness include pulmonary edema (cardiogenic and noncardiogenic), pneumonia, interstitial fibrosis or inflammation, tumor, and atelectasis. As noted above, bronchospasm not only causes increased flow resistance but simultaneously worsens the elastic load by augmenting PEEPi.
The chest wall includes the thorax, diaphragm, and abdomen. Therefore, obesity, rib fracture, pneumothorax, pleural effusion, ascites, chest wall abnormalities, and abdominal distention (see Chap. 58) contribute to the work of breathing. These factors are particularly relevant in the postoperative setting.
Dividing the work of breathing into resistive and static components is a useful way of analyzing the effort of each breath. However, even if the work of each breath remains constant, an increase in respiratory rate increases the load. Increased minute ventilation (V.e) requirements can be divided into those due to excess carbon dioxide production (˙VCO2) and those from worsened dead space. The first category includes excessive caloric intake, fever, agitation, muscular exertion (including shivering and respiratory distress) and hypermetabolism due to injury or infection. New dead space may be caused by pulmonary embolism, hypovolemia, PEEP (including PEEPi), or shallower breathing (which raises the dead space fraction, Vd/VT). Partitioning of the lung mechanics into resistive and static components in the intubated COPD patient may be inaccurate if the end-inspiratory pause maneuver is too short and if there is rapid spontaneous respiratory effort.86
MULTIFACTORIAL RESPIRATORY FAILURE
In order to analyze respiratory failure in a way that facilitates management, it is important to dissect the very complex real-life patient into simple components of deranged neuromuscular competence and load. However, the patient with ACRF due to isolated rib fracture or hypokalemia is uncommon. More often, numerous contributors to both decreased strength and increased load are implicated. For example, a patient admitted with pneumonia and worsening bronchospasm may also have hypophosphatemia, heart failure, and malnutrition. Further, patients with COPD are significantly more likely to suffer from comorbid cardiovascular and cerebrovascular disease and diabetes mellitus,87 all of which can contribute to and compound acute ACRF in COPD. Aspiration at the time of intubation, abdominal distention related to attempts to feed enterally, and overzealous parenteral nutrition complete the picture of multifactorial ACRF. Although the situation in such a patient may seem overwhelmingly complex, we caution against a “shotgun” approach to evaluation and management of patients with multifactorial respiratory failure. Despite the temptation to provide all possible interventions for these very ill patients, a systematic approach to each component of respiratory failure leads logically to a plan for treatment.
Failure to aggressively treat mild to moderate acute exacerbations of COPD prior to the development of ACRF and other organ failure is associated with an increased risk of emergency hospitalization and delayed recovery.88 We generally urge caution in extrapolating clinical data from studies of respiratory failure complicating acute status asthmaticus to ACRF from COPD; the time course, biological mechanisms of bronchospasm and airway inflammation as well as propensity for reversibility differ between these conditions. However, it is important to appreciate that up to a third of patients with ACRF have underlying reversible airway obstruction consistent with the diagnosis of asthma and that a tailored approach is required for those patients. The unequivocal role of noninvasive positive pressure mechanical ventilation (NIV) justifies its position as the cornerstone in the therapeutic approach to critically ill patients with ACRF (see Chap. 44). We consider NIV to be the “bookends” in the therapeutic library of therapy for ACRF, providing firm support on either end of an exacerbation of ACRF. This approach is discussed below but incorporates early initiation of NIV to avoid MV and improve survival and later use of this tool for early liberation from MV. We describe below three phases of management of the ACRF patient: early ACRF, late ACRF requiring intubation for MV, and liberation from the ventilator.
Phase 1: Early ACRF: The goals of management in the patient not yet intubated are to avoid mechanical ventilation when that is possible and to recognize progressive respiratory failure when it is not. The proven efficacy of NIV to decrease mortality, avert the need for intubation, reduce complications—specifically hospital-acquired pneumonias—and shorten duration of hospital stay with a favorable health economic impact makes this therapy one of the most important developments in the management of these patients, because it buys time for the physician to treat precipitants of ACRF and for the patient to improve.
Current guidelines recommend NIV as definitive and first-line therapy for COPD-related ACRF,10,89,90 particularly in patients with persistent hypercapnic ventilatory failure despite optimal medical therapy. Most centers administer NIV via a full face91 or nasal mask and pressure-support ventilation, although helmet interfaces, poncho wraps, and other ventilator modes can support NIV. For the dyspneic ACRF patients who preferentially mouth breathe, a full-face mask is preferable to minimize oral air leakage. The full-face mask also requires less patient cooperation than the other interfaces. A tight-fitting mask allows substantial ventilatory assistance yet provides for brief periods off of the ventilator during which patients can speak, inhale nebulized medications, expectorate, and swallow liquids.
NIV has been systematically evaluated in several large studies. The outcomes from these studies have been synthesized by several groups.92,93 A Cochrane meta-analysis of 14 randomized controlled studies of NIV versus usual medical care (UMC) included a total of 758 patients with ACRF.92 Age ranged from 63 to 76 years, admission pH 7.26 to 7.34, admission PaCO2 57 to 87 mm Hg, admission PaO2 39 to 73 mm Hg and FEV1 0.68 to 1.03 L. The eight largest studies enrolled 40 or more patients. Five studies were conducted in an ICU setting and the remainder in medical wards or progressive care units. Mean duration of NIV was 4.3 days (range, 3-10 days). The combined analysis demonstrated that: Treatment failure was less likely with NIV than UMC (RR 0.48; 95% CI 0.37, 0.63) with an NNT of 5 (95% CI 4, 6) and mortality was reduced by 48% (RR for death 0.52; 95% CI 0.35, 0.76; NNT 10 (95% CI 7, 20). Notably, the mortality reduction was evident regardless of whether ACRF was treated in an ICU or in a general ward. Additionally, there was a 60% reduction in requirement for intubation with NIV, and rapid improvements in respiratory rate, PaCO2, and pH. Hospital length of stay was reduced by 3.24 days although the trend to reduced ICU LOS (4.71 fewer days) did not reach significance. Interestingly, the improvement in PaO2 was heterogeneous between studies and was not significantly different at 1 hour after initiation of treatment. The finding of another meta-analysis revealed essentially similar findings for survival and clinical improvement, but subgroup analysis suggested that the benefit was limited to patients with severe but not mild ACRF. This hypothesis has not been tested in a prospective, stratified fashion.94
NIV has been used for prolonged periods (more than 1 week) and has been shown to relieve symptoms, reduce respiratory rate, increase tidal volume, improve gas exchange,95 and lessen the amplitude of both the diaphragmatic electromyogram and the transdiaphragmatic pressure.96 Complications of the mask have been minor and few; local skin breakdown has been attributed to the tight-fitting mask but can be avoided by applying a patch of wound care dressing. Only a few patients cannot tolerate face or nasal masks, and some of these patients respond to judicious and carefully monitored use of anxiolytics. Aspiration of gastric contents has only rarely been noted in these patients, even when a nasogastric tube is not routinely placed; however, impaired mentation probably increases this risk. However, in selected patients with severe hypercapnic encephalopathy but stable hemodynamics, NIV may be as effective as in less cognitively impaired patients, although predictably, those patients require higher levels of inspiratory pressure support and prolonged ICU stays.97
Careful patient selection is essential for successful NIV in ACRF and is summarized in Table 54-2.98 Although a nasal mask is objectively as effective and well tolerated as a full-face mask,99 we typically begin therapy with a full-face mask. Gel-insert masks are preferable in an effort to prevent skin breakdown of the nasal bridge. Claustrophobia is quite frequent on initial application and may be effectively circumvented by enlisting the patient (if capable) to hold the mask lightly in position without securing the head-straps. Additionally, we attempt to use 3 to 5 cm H2O expiratory positive pressure (EPAP) only for the first several minutes. Thereafter, inspiratory positive pressure (IPAP) of 5 to 8 cm H2O is delivered using a pressure limited mode on a noninvasive ventilator until the patient is able to tolerate the mask comfortably and synchronize with the ventilator.100 After applying head-straps, we aim to achieve an EPAP of 2 to 5 cm H2O (to counterbalance PEEPi) and an IPAP can be increased by 2 to 5 cm H2O every 10 minutes, to a target of 15 to 18 cm H2O (equivalently, 2-5 cm H2O PEEP with 13-16 cm H2O pressure support) to assist alveolar ventilation or patient tolerability has been reached.89,101 Higher pressures can sometimes be used, but they tend to be limited by air leak or mask discomfort. The PEEP component of NIV is important102 and does not usually cause incremental hyperinflation.103 Indeed, continuous positive airway pressure (CPAP) alone (without ventilatory assistance) reduces the work of breathing, improves gas exchange, leads to subjective benefit, and sometimes can avert intubation104-106 when applied to patients with ACRF. When PEEP was added to pressure-support ventilation in ventilated patients with COPD, inspiratory effort fell another 17%, and patient-ventilator synchrony improved.79
Selection Criteria for NIV for ACRF
| Selection Criteria | |
|---|---|
| Inclusion Criteria | Exclusion Criteria (any may be present) |
|
|
The use of helium-oxygen mixtures as a driving gas for NIV has been considered attractive because of theoretically beneficial alterations in gas density and reduction in resistive pressure gradients across the inflamed and mucus impacted respiratory tree. However, in 204 patients with AECOPD requiring NIV for severe dyspnea and hypercarbia (PaCO2 >45 mm Hg), use of heliox (35% FiO2) as a driving gas was no less likely to be associated with requirement for intubation than air:oxygen mixture with the same FiO2.107
NIV is not uniformly successful in patients with ACRF.108 Essential to successful patient-NIV synchrony are trigger settings for inspiration, pressurization rates (rapid pressurization reduces diaphragm work but results in increased leak) and inspiratory-to-expiratory cycling (mask leaks can result in delayed cycling). These aspects are reviewed in detail in Chap. 44. A salutary response is typically evident within 10 minutes of beginning NIV, as indicated by a falling respiratory rate and heart rate as well as by the patient’s subjective assessment. Occasional patients feel claustrophobic and may show objective worsening with NIV. Although we occasionally use pharmacologic anxiolytic therapy with success, this course has obvious attendant risks and should only be undertaken with appropriate safeguards.
A risk of NIV that is not often discussed is its potential to lull the physician-nurse-respiratory therapist team into a sense of comfort while the patient continues to worsen. Time spent trying NIV will, in these patients, potentially lead to a later, more urgent intubation in a more exhausted patient with significantly greater tissue hypoxia. This very real concern was corroborated by Chandra et al who reported an alarming increase in the frequency and mortality rates for ACRF patients requiring invasive MV after a trial of NIV—presumably because NIV was ineffective. Over the decade from 1998 to 2008 in the USA, approximately 5% of patients with AECOPD required transition to MV from NIV. However, mortality for this group increased progressively at a time when mortality for NIV alone patients was decreasing. By 2008, patients transitions to MV had a 61% higher odds of death compared with patients initiated on MV (95% CI 24%-109%) and startling 677% greater odds of death compared with patients treated with NIV alone (95% CI 475%-948%).8
Avoiding intubation and conventional mechanical ventilation nearly always depends on discerning the cause of ACRF and reversing it. Thus, while NIV is initiated, each of the potential causes enumerated in Figure 54-2 should be reviewed in light of the clinical presentation. On occasion, impending respiratory failure can be averted by a specific intervention targeted to one precipitant, such as rib fracture (intercostal nerve block) or pulmonary edema (diuresis, afterload reduction). More often, several contributors are identified, such as worsened bronchospasm, electrolyte derangement and infection. Thus treatment must be broad based.
Since NIV is intermittently but variably interrupted for patient comfort and nutritional intake, the approach to weaning and liberation from NIV has not been prospectively evaluated and requires an individualized but consistent approach. Weaning to liberation can be initiated if the patient feels symptomatically improved, pH is >7.35 and stable, there is progress toward resolution of underlying precipitants and respiratory rate during spontaneous breathing has normalized following the first 24 hours or longer. We find that initially reducing IPAP levels by 10% to 20% over 12 hours while retaining EPAP at 7 to 10 cm H2O is an effective first step. Many patients with significant PEEPi experience increased work of breathing if EPAP is reduced too rapidly. We provide periods of 30 to 60 minutes of spontaneous breathing with supplemental nasal cannula oxygen alternating with NIV for 3 to 4 hours. Periods of spontaneous breathing are progressively prolonged over 1 to 2 days until the patient is tolerating 4 to 6 hours off NIV. In up to a third of patients, the initial efforts to liberate can be unsuccessful. If rapid shallow breathing, pursed-lip breathing, or tachycardia recurs, our approach is to defer liberation efforts for a further 8 to 12 hours and continue NIV or make a decision to advance to intubation (see below). Guideline recommendations suggest continuing NIV for 16 hours on day 2, 12 hours on day 3 including 6 to 8 hours overnight use, and discontinuing NIV on day 4, unless continuation is clinically indicated.89,109 Our preference is to provide NIV at night for the first 2 to 3 nights after the patient is weaned.
Oxygen Supplemental oxygen administration is a cornerstone of treatment.110 Despite this a pervasive myth that patients with ACRF rely on hypoxic drive to breathe persists. Physicians are often hesitant to supply oxygen, fearing that patients will stop breathing, necessitating intubation. Since these patients are typically hypoxemic on presentation (PO2 usually about 30-40 mm Hg), failure to supply adequate oxygen is a potentially devastating treatment error. Unrelieved hypoxemia in the face of acidemia, fatiguing respiratory muscles, and an often failing right ventricle risks arrhythmia, myocardial infarction, cerebral injury, renal failure, and respiratory arrest.
Related posts:







Full access? Get Clinical Tree


