KEY POINTS
Current paradigms of sepsis include both pro- and anti-inflammatory pathway activation to different degrees and at different phases of the syndrome.
Failure to recognize and understand the dynamic changes in immune response in sepsis may in part explain the failure of a number of anti-inflammatory drugs and biologics studied in critically ill patients.
The anti-inflammatory or immunosuppressed state associated with sepsis and other forms of critical illness is often protracted and places patients at risk for complicating nosocomial infections and activation of latent infections.
When clinically significant, the anti-inflammatory state associated with sepsis is termed immunoparesis or immunoparalysis.
Cell and humoral biomarkers are needed to properly characterize the individual patient’s immune status to guide targeted and personalized therapy to modulate both excessive immune stimulation as well as immune suppression.
INTRODUCTION
Sepsis [σήψις] is the original Greek word for the “decomposition of animal or vegetable organic matter in the presence of bacteria.” The word is found for the first time in Homer’s poems, where Sepsis is a derivative of the verb form sepo [σήπω], which means “I rot.” The term sepsis is also found in the Corpus Hippocraticum exchangeably with the word sepidon [σηπεδών] (“the decay of webs”): Epidemic. B. 2,2, Prorret. I. 99. Aristoteles, Plutarch, and Galen use the word sepsis [σηψις] in the same meaning as Hippocrates.1
This original meaning connoted decay and wound putrefaction and described a process of decomposition of organic matter and tissue breakdown resulting in disease (foul odor, pus formation, dead tissue) and eventually to death.2 Thus, the word sepsis has persisted for 2700 years with more or less unchanged meaning. Subsequent works just confirmed the causal link between microbes and suppurative infections or systemic symptoms and clinical findings from infections establishing the infections as the underlying disease. Hugo Schottmuller in 1914 founded the modern definition of sepsis and was the first to describe that the presence of an infection was a fundamental component of the disease.3
In 1972, Lewis Thomas described sepsis in the following way: “It is our response to [the microorganism’s] presence that makes the disease. Our arsenals for fighting off bacteria are so powerful … that we are more in danger from them than the invaders.” and popularizing the theory that “…it is the [host] response … that makes the disease.”4 Finally, the concept entered into daily clinical practice when Roger Bone and colleagues defined sepsis as a systemic inflammatory response syndrome that can occur during infection.5
In recent years this syndromic characterization of sepsis has been expanded to SIRS (systemic inflammatory response syndrome), CARS (compensatory anti-inflammatory response syndrome), and MARS (mixed antagonists response syndrome), with recognition that immune dysfunction during sepsis may be a significant aspect of pathogenesis.6,7
Currently sepsis is considered a host immune response to infection, which clinically results in a continuum of disease categorized as sepsis, severe sepsis, septic shock, and multiorgan failure (MOF). Also, sepsis is the maladaptive immune response of the host to invading pathogens in normally sterile sites of the body. In severe sepsis and septic shock this inappropriate immune response to infection leads to mismatch of host response to the pathogenic stimuli so profound as to finally lead to cellular dysfunction and ultimately to organ injury and dysfunction or failure.
The immune profile of this host-pathogen mismatch can be predominately proinflammatory (systemic inflammatory response syndrome, SIRS), mixed (mixed antagonistic response syndrome, MARS), or anti-inflammatory (compensatory anti-inflammatory response syndrome, CARS). The final result is various degrees of hyperinflammation, immunosuppression, abnormal coagulation, and microcirculatory dysfunction, all which may contribute to organ injury and cell death.2,6
Clinical diagnosis of severe sepsis or septic shock although valuable and of significant importance for the management of septic patients may lead to extremely heterogeneous cohorts in terms of patients’ immunological status. This heterogeneity offers one explanation for the failure of prior trials of biologic therapies for sepsis, since treatments that focused on attenuating the initial inflammatory response of sepsis in a sense ignored and in fact might have exacerbated the progressive development of immunosuppression in some patients.8-11
Immune status characterization during the course of sepsis may identify patients who could benefit from immunotherapy tailored to their particular circumstances. These patients may be those who develop septic shock and die early from multiorgan failure or those who develop late immunosuppression after surviving the initial septic shock but fail to completely recover from persisting sepsis syndrome. The latter patients often develop what appears to be chronic sepsis, with recurrent nosocomial infections and eventual recurrent and refractory septic shock. In a sense these patients may be considered to have yet another organ system failing in the face of sepsis—their immune system.
Sepsis is a major health care problem due to the high morbidity and mortality of the syndrome, which has very high health care costs. Despite intense research and recent advances in treatment, mortality remains extremely high, reaching 40% to 60% in high-risk patient populations.
Infections caused by diverse microorganisms and involving many different body sites may present as SIRS, which is a clinical syndrome defined by (a) hyperthermia >38.0°C or hypothermia <36.0°C, (b) tachycardia (heart rate >90/min), (c) tachypnea (respiratory rate >20 per minute) or hyperventilation (<SPAN role=presentation tabIndex=0 id=MathJax-Element-1-Frame class=MathJax_Error style="POSITION: relative" data-mathml='PaCO2′>[Math Processing Error]PaCO2 <32 mm Hg), (d) leucocytosis (WBC count >12.000/mm3) or leukopenia (WBC count <4.000/mm3 or the presence of >10% immature neutrophils (bands) as defined by the American College of Chest Physicians/Society of Critical Care Medicine (ACCP/SCCM) Consensus Conference.12 SIRS driven by infection progresses along a continuum, described as sepsis, severe sepsis, septic shock, and multisystem organ failure. Along this continuum the host’s immune system is operating at varying levels of activation, driven by complex interactions between the host and infectious agent(s). Host immune response includes innate immune response that incorporates humoral and cellular components. The humoral component includes release of cytokines, chemical substances that are directly toxic to invading microbes or that act as mediators for immune cell activation. The cellular component includes circulating monocytes, tissue macrophages, neutrophils, and lymphocytes.
As a result of the actions of the innate immune system tissue macrophages engulf and digest pathogens, produce cytokines, and present pathogen particles (antigens) to lymphocytes, providing linkage to the adaptive immune system. Neutrophils are attracted by chemokines and migrate to infected tissues where they phagocytose pathogens and secrete toxic substances such as reactive oxygen species (ROS) that destroy invading microorganisms. Eosinophil and basophil granulocytes secrete mediators creating an inflammatory milieu locally in the infected tissues and systemically in the circulation. As a consequence peripheral leukocytosis is observed due to bone marrow stimulation with left shift of neutrophils (immature forms), dilation and leakage of the adjacent vessels due to the action of vasoactive inflammatory mediators (NO) to facilitate the migration of inflammatory cells into the infected tissue, which leads to efflux of plasma into tissues. Taken all together these processes lead to clinical signs of local inflammation, including redness (rubor), swelling (tumor), increased temperature (calor), and pain (dolor).
Thus, infection may present with signs and symptoms of SIRS and may resolve with the use of antibiotic and/or other supportive measures. Normally, the immune system controls local inflammation and eradicates invading pathogens. When local control mechanisms fail, however, systemic inflammation and then sepsis occurs.
Cells of the innate immune system recognize molecular patterns of most microbes including viruses, bacteria, fungi, and protozoa to produce inflammation at the local level or systemically. Thus inflammation starts when damage-associated molecular patterns (DAMPs) bind to immune cell pattern recognition receptors (PRRs), which rapidly initiate host defense responses. DAMPs are both pathogen-associated molecular patterns (PAMPs) that are expressed by both invading and innocuous microorganisms and intracellular proteins or mediators that are released from damaged tissues and dying cells, which are known as alarmins such as high mobility group box 1 and S100a proteins. PAMPs include lipopolysaccharides (LPS, endotoxin) contained in the cell wall of gram-negative bacteria, lipoteichoic acid and peptidoglycan from gram-positive bacteria, bacterial DNA, or viral RNA. PRRs include Toll-like receptors (TLRs), intracellular NOD proteins, and peptidoglycan recognition proteins.
The recognition, binding, and interaction of DAMPs (eg, LPS) by PRRs (eg, TLRs) located on the immune cell surface result in signal transduction and in turn to a complex intracellular cascade of enzymes (kinases), which activate proteins. These proteins activate additional intracellular pathways leading to activation of transcription factors within the cell nucleus binding to DNA, thus activating hundreds of specific genes coding for proteins, which are increased during the inflammatory process in a time-dependent fashion. For example, in gram-negative sepsis LPS binds to TLR4 and CD 14 activating myeloid differentiation protein (MyD)-88, which then activates interleukin-1 receptor–associated kinase (IRAK), which, in turn, stimulates the tumor necrosis factor receptor–associated factor (TRAF) and, consequently, the TRAF-associated kinase (TAK). As a result, the nuclear transcription factor, nuclear factor kappa B (NFκB), is liberated from its inhibitor (IκB) and is able to dislocate into the cell nucleus and bind to DNA and modulate gene function.13-15
During sepsis high levels of circulating DAMPs from invading microorganisms and/or damaged host tissue activate host immune cells, leading to inflammation characterized by the so-called cytokine storm. The early phase of sepsis creates a proinflammatory environment, which is caused by the excessive activation of the host immune system by tissue damage and/or severe infection, leading to severe dysregulation of various body systems.16 Central hubs of the inflammatory response during sepsis include the complement anaphylatoxin C5a, macrophage migration-inhibitory factor (MIF), Toll-like receptor 4 (TLR4), high-mobility group box 1 protein (HMGB1), interleukin-17A (IL-17A) but also the coagulation, the endocrine, the innate and adaptive immune, and the autonomic nervous systems (adrenergic and cholinergic pathways).3
One of the significant molecules produced during sepsis is TNF, which propagates inflammatory pathways in multiple organ systems and also plays a very important role in the activation of programmed cell death or apoptosis. Also, interleukin (IL)-6 induces the production of acute phase proteins in the liver, for example, C-reactive protein and fibrinogen. Another enzyme activated during sepsis is inducible nitric oxide synthase (iNOS) leading to nitric oxide (NO) production and finally cyclic guanosine monophosphate (cGMP) that leads to local and systemic vasodilation, which correlates clinically to hypotension and shock.17
Vasodilation and intravascular volume depletion from increased capillary leak and external losses observed in early sepsis lead to underfilling of the heart and a low cardiac output, which in conjunction with myocardial depression potentially causes an oxygen supply-demand imbalance in various organ beds. Further imbalance may occur due to decreased oxygen delivery to the tissues by alterations of the microcirculation observed in patients with sepsis.18 Following adequate volume resuscitation patients typically exhibits high cardiac output hypotension, although during the early hours to days of sepsis a propensity for continued loss of intravascular volume persists often resulting in recurrent hypovolemia and requiring the clinician managing the patient with septic shock to repeatedly return to the question of whether additional intravascular volume is needed.
Also, the inflammatory insult of sepsis appears capable of causing structural and functional damage to the mitochondria.19,20 Mitochondrial dysfunction may be due to direct inhibition of the respiratory enzyme complexes from increased concentrations of nitric oxide and its metabolite, peroxynitrite, and by direct damage from increased production of reactive oxygen species. Also, recent studies report a genetic downregulation of new mitochondrial protein formation, which is associated with intramitochondrial defense mechanisms (glutathione, superoxide dismutase) being depleted or overwhelmed.21,22
The therapeutic window during this initial hyperinflammatory response for initiating treatment with anti-inflammatory drugs is likely narrow (<24 hours), after which a treatment to increase immune function may be more beneficial. This may in part explain a number of negative therapeutic trials directed at reducing inflammatory mediators in septic patients. Evidence from several studies has shown that certain anti-inflammatory pathways seem activated very early in septic patients.23-25 The systemic anti-inflammatory response may be useful for the attenuation of deleterious systemic proinflammatory effects and the concentration and compartmentalization of the inflammation at the site of infection.26
However, when anti-inflammatory mechanisms dominate, the immune system is depressed, a condition termed immunoparesis or immunoparalysis, and the body’s susceptibility to nosocomial infections and the reactivation of dormant pathogens such as cytomegalovirus is increased.27,28 The state of immunoparesis is associated with declining levels of numerous hormones, a reduced metabolic rate, and in some tissues frank bioenergetic failure. The observation of these responses to infection has raised the hypothesis that immune system stimulation during this phase of sepsis could be beneficial.29,30 Similar to the notion of SIRS, this phase of the course of sepsis has been termed the compensatory anti-inflammatory response syndrome (CARS). Interestingly, it seems most deaths related to sepsis occur during this phase.
FIGURE 62-1
Evolution from infection to septic shock and immunoparalysis. DAMPs, damage-associated molecular patterns; LPS, lipopolysaccharide; PAMPs, pathogen-associated molecular patterns; PRRs, pattern recognition receptors; RRT, renal replacement therapy; SIRS, systemic inflammatory response syndrome; TLRs, Toll-like receptors.
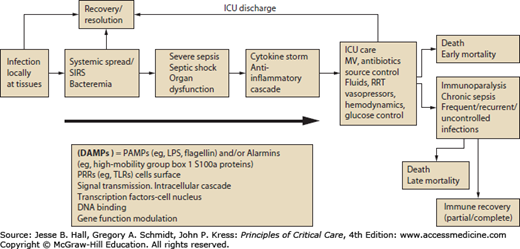
As described above, much of our therapy for sepsis is “early and goal directed” and unfolds in the early phase of the syndrome during which large and uncontrolled release of endogenous and exogenous mediators of inflammation occurs. Despite our efforts to identify patients early and deploy therapies such as early fluid therapy, mortality remains as high as 30% to 40% for the highest risk patients.32-34 Much of this mortality occurs during the state of immunoparesis, related to “second and third hits” in the form of nosocomial superinfections.34
During this phase of sepsis activation of anti-inflammatory mediators such as interleukin 10 (IL-10), and transforming growth factor-beta (TGF-β) are well described, and it appears these pathways are activated in the course of strong proinflammatory stimulation, perhaps as an effort to achieve homeostasis. Modification of cellular function is another feature of this state of immunoparesis, and sustained periods of monocyte deactivation characterized by defective phagocytosis, altered antigen presentation, and reduced production of inflammatory cytokines by these cells have been described.25
This reprogramming event of immune function occurs at other levels as well. At the transcriptional level, downregulation of genes encoding proinflammatory cytokines and other acute phase proteins is accompanied by an outpouring of anti-inflammatory cytokines and cytokine inhibitors, and downregulation of cytokine receptors.35 Neuroendocrine response also appears to attenuate inflammatory cytokine synthesis and also results in reduction of antigen expression on antigen presenting cells.25,36 In addition, specific subsets of lymphocytes, dendritic cells, and epithelial cells undergo apoptosis at extremely accelerated rates after a typical septic stimulus in patients.37,38 Uptake of apoptotic cells further impairs host immunity by inducing an anti-inflammatory phenotype in phagocytic cells that consume the cellular corpses.39 Prevention of this sepsis-induced apoptosis apparently attenuates the immunosuppressive cascade and leads to sustained immunity.37
T-regulatory cells and myeloid-derived suppressor cells are also activated during the later phases of sepsis and appear to participate in general downregulation of immune response and the inflammatory state.39-42 Other described mechanisms of this immunosuppression include massive apoptosis-induced depletion of lymphocytes and dendritic cells, decreased expression of the cell-surface antigen–presenting complex HLA-DR, and increased expression of the negative costimulatory molecules programmed death 1 (PD-1), cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4), and B- and T-lymphocyte attenuator (BTLA) and their corresponding ligands (eg, PD-1 ligand [PD-L1]). Furthermore, the numbers of regulatory T cells and myeloid-derived suppressor cells (MDSCs) are increased, and there is a shift from a phenotype of inflammatory type 1 helper T (Th1) cells to an anti-inflammatory phenotype of type 2 helper T (Th2) cells characterized by the production of interleukin-10.
The net result is a severely compromised innate and adaptive immune system with poorly functional “exhausted” CD8 and anergic CD4 T cells. Targets of potential immunotherapeutic approaches (see Fig 62-1) include agents that block apoptosis, block negative costimulatory molecules, decrease the level of anti-inflammatory cytokines, increase HLA-DR expression, and reactivate “exhausted” or anergic T cells. FLT-3L denotes Fms-related tyrosine kinase 3 ligand, GM-CSF granulocyte–macrophage colony-stimulating factor, LFA-1 lymphocyte function–associated antigen 1, and tumor necrosis factor α (TNF-α).
In this later, hypoimmune phase of the disease, hemodynamics will most likely be relatively stable. Mechanical ventilation is often necessary, and a requirement for renal replacement therapy is common. The main focus of the treating clinician will now be on the prevention and treatment of secondary infections, such as ventilator-associated pneumonia, catheter-related infections, and fungal infections. The patient will most likely be treated with various antibiotic and antifungal regimes, and multiresistant strains may be detected in the further course of the disease.
In response to stress and injury (of which septic shock is a typical example), the body develops compensatory mechanisms to prevent systemic inflammation. This anti-inflammatory response is a homeostatic mechanism that occurs in all patients. It protects against lethal overwhelming inflammation during the first hours of the syndrome, but becomes deleterious as it persists because nearly all immune functions are compromised. As all cell types—neutrophils, monocytes/macrophages, and lymphocytes—are impaired, both innate and specific immunities are depressed. The terms immunoparalysis and immunosuppression have been proposed to describe the global incapacity of the body to mount any kind of immune response.43
In many cases of sepsis, the immune system fails to eradicate the infectious pathogens, and a prolonged phase of sepsis-induced immunosuppression begins, characterized by a failure to eradicate the primary infection and by development of secondary nosocomial infections.
It seems that the majority of shock-related deaths occur during this secondary hypoimmune state. It has been proposed that patients who survive the initial hyperinflammatory response but subsequently die from sepsis are those who do not recover immune function.23,25,44-48
The clinical relevance of this immunosuppressed state is evidenced by the frequent occurrence of infection with relatively avirulent and often multidrug-resistant bacterial, viral, and fungal pathogens (Stenotrophomonas, Acinetobacter, and Pseudomonas spp, enterococci, cytomegalovirus, herpes simplex virus, and Candida spp).35,49-51
This more protracted phase of sepsis is associated with increased length of stay, significant morbidity and mortality, and increased costs.23,25,51-53
Two recent studies in human subjects shed light on one of the most important and relatively underrecognized mechanisms of sepsis immunopathology. They provide evidence that the otherwise dormant viruses cytomegalovirus (CMV) and herpes simplex virus (HSV) are reactivated in critically ill individuals—adding strength to the concept that a key aspect of critical illness is immunosuppression. Limaye et al28 examined the incidence of reactivation of CMV in 120 CMV-seropositive critically ill individuals, many of whom apparently had sepsis. Before their illness, these people had normal immunity. CMV viremia, as assessed by real-time PCR, occurred in 40 subjects (33%), indicating that CMV reactivation occurs frequently in the critically ill. CMV reactivation was associated with prolonged stay and death. These findings dovetail with an earlier study by Luyt et al who reported a 21% incidence of HSV bronchopneumonitis that was attributed to viral reactivation in critically ill, immunocompetent individuals requiring prolonged mechanical ventilation.28,54 These studies show that critically ill individuals who had normal immunity before hospitalization become profoundly immunocompromised during a protracted illness, thereby enabling reactivation of latent viruses that may become clinically relevant.
CARS-associated immunosuppression may persist for days, weeks, months, or years following the initial SIRS event.55 This long-term immunosuppression manifests in patients as a significant decrease in 1- and 8-year survival for patients with severe sepsis compared to age-matched controls, often due to infections. The poor survival persisted when multivariable analysis was conducted to adjust for prehospitalization comorbid conditions.56,57
Also, survivors of septic shock are vulnerable to A fumigatus and P aeruginosa long after sepsis-induced proinflammatory immune response has been resolved, indicating that CARS exists as a long-term phenomenon, which requires its own diagnostic tools and therapies for the patients to survive.55 Several other studies have also supported the finding that mortality remains high for several years among hospital survivors of infectious diseases and sepsis.58-63 When multivariable analyses were used to compare long-term mortality estimates, with and without chronic health conditions, the long-term estimates remained unchanged. Thus, the influence of chronic health conditions on long-term survival is small and that the acute illness is more likely to contribute to increased long-term mortality.63
Three potential outcomes are possible following hospitalization for infection and sepsis. Approximately one-fourth of severe sepsis patients die during the hospitalization. The remaining patients who are discharged alive either recover completely or experience incomplete recovery. Those with incomplete recovery are at increased risk of subsequent acute illnesses, which eventually leads to death.63 Several lines of evidence suggest that consequences of severe sepsis could be due to immune suppression. Although the exact mechanisms for protracted increased susceptibility to infections is unclear, impaired neutrophil function appears to play an important role.64
Another study showed that mHLA-DR is an independent predictor of mortality in septic shock patients. Being a marker of immune failure, low mHLA-DR may provide a rationale for initiating therapy to reverse immunosuppression.65 In septic shock patients, after adjustment with usual clinical confounders (including need for mechanical ventilation and central venous catheterization), persistent low mHLA-DR expression remained independently associated with the development of secondary nosocomial infections (NI).66
Related posts:






Full access? Get Clinical Tree


