KEY POINTS
Pulmonary embolism (PE) is common and potentially lethal, yet readily treatable.
Prophylaxis and accurate diagnosis are essential to improving outcome.
The cause of death in PE is most often circulatory failure (acute cor pulmonale) due to right heart ischemia.
There is no perfect diagnostic test for PE; accurate diagnosis requires both an informed clinical pretest probability and a stepwise application of helical CT angiography and/or LE duplex.
A careful risk assessment may identify patients ideal for outpatient therapy. Conversely, patients with hypotension or right ventricular strain are at significantly higher risk for death from PE, and warrant ICU admission.
While low-molecular-weight heparin (LMWH) is approved and recommended as the initial therapy for PE, critically ill patients often have reason for a shorter-acting medication. Unfractionated heparin is typically used to maintain the partial thromboplastin time (PTT) at 1.5 to 2.5 times control.
Thrombolytic therapy is lifesaving and possibly in those with isolated RV dysfunction in patients with massive embolism and circulatory instability, but does not seem beneficial in patients without shock.
Air and fat embolism usually present as acute respiratory distress syndrome (ARDS), and are managed with mechanical ventilation, oxygen, and positive end-expiratory pressure (PEEP).
This chapter covers diseases involving embolism to the pulmonary circulation, including pulmonary thromboembolism, as well as the less common conditions of venous air embolism and fat embolism. Thromboembolism is predominantly an acute circulatory insult, with important but less dramatic consequences for gas exchange. In contrast, both air and fat embolism usually present as acute hypoxemic respiratory failure (AHRF). All three of these forms of embolism may cause acute right heart failure, more fully discussed in Chap. 38.
PULMONARY THROMBOEMBOLISM: EPIDEMIOLOGY IN THE ICU
PE is a dramatic and life-threatening complication of underlying deep venous thrombosis (DVT). Therefore, much of the management of PE is grounded in the prophylaxis, diagnosis, and treatment of DVT. While extensive prospective data regarding the diagnosis and treatment of PE are available, the vast majority of patients in such trials have not been critically ill, and thus the treatment for ICU patients with thromboembolic disease relies on extrapolation, and may lack the strength of evidence now available for most patients with PE or DVT. Nonetheless, important distinctions exist between the critically ill and noncritically ill patient populations when considering PE diagnosis and treatment.
Pulmonary thromboembolism is a common illness, which accounts for substantial morbidity and mortality. Interesting trends have been recently reported regarding the incidence of PE. Historically, acute PE was believed to be frequently underdiagnosed, and a frequent cause of unexplained sudden death, as it was estimated that up to 25% of patients may die before admission.1 However, a recent time-trend analysis using an administrative database demonstrated a dramatic increase in PE incidence in the United States, from approximately 62 cases per 100,000 population to over 113 cases per 100,000.2 This change in the incidence of PE was coincident with the introduction and adoption of multidetector row computed tomographic scanning as a primary diagnostic modality for PE, raising the possibility that PE is now overdiagnosed, given that across the same period, the mortality rate was unchanged and case fatality rates fell.2 The use of the term overdiagnosis remains controversial as small and even asymptomatic PEs can recur, albeit at apparently much lower rates.3,4 Failure to diagnose symptomatic PE remains a serious management error since it has been estimated that over 30% of untreated patients die, while only 8% succumb with effective therapy.5-7
ICU patients are an inhomogeneous group of patients and the incidence of thromboembolism ranges widely across subsets of patients. For general medical/surgical ICU patients, the incidence of DVT is approximately 30% based on screening studies.8,9 As discussed below, prophylaxis of these patients lowers the risk of VTE and has become standard practice in most units; a large registry of hospitalized patients with DVT found that the majority had not received prophylaxis prior to their DVT diagnosis.10 Additional risk factors, discussed in more detail below, include advancing age, malignancy, and presence of an indwelling venous catheter or pacemaker. Many ICUs now protocolize prophylaxis decisions in an effort to increase adherence to prophylaxis guidelines.
In spite of great strides in the understanding of VTE, pulmonary embolism (PE) continues to cause substantial morbidity and mortality. Critically ill patients form a unique and challenging subset of those at risk. The presence of indwelling lines and forced immobility make these patients particularly susceptible to venous thromboemboli. Diagnosis, which is difficult even in ambulatory patients, is further impeded by barriers to communication and physical examination. Moreover, alternate explanations for hypoxemia, lung infiltrates, respiratory failure, and hemodynamic instability are readily available, such that a diagnosis of pulmonary thromboembolism may not be considered likely. Finally, critically ill patients are likely to have limited cardiopulmonary reserve, so that pulmonary emboli may be particularly lethal.
Venous thrombosis begins with the formation of microthrombi at a site of venous stasis or injury. Thrombosis impedes flow and generates further vascular injury, favoring progressive clot formation. In some patients, clot becomes substantial and propagates to a proximal vein where it has the potential to embolize to the pulmonary circulation. Thrombus in the vasculature causes chiefly a mechanical obstruction to flow, but also triggers the release of vasoactive and, for PE, bronchoreactive substances like serotonin which can exacerbate ventilation—perfusion mismatch.
Most clinically relevant pulmonary emboli originate as proximal venous thrombi in the leg or pelvic veins. However, in the ICU, the routine placement of upper body catheters for vascular access, monitoring, drug administration, and nutrition raises the likelihood of important upper body sources of thrombi. Up to one-third of patients with indwelling venous catheters have ultrasound-detectable clot at screening, although in one study none were symptomatic, and none developed a symptomatic pulmonary embolus over 18 months.11 Subsequently, in a large international registry of patients with clinically diagnosed VTE, the proportion of upper extremity DVT was low (4%) and patients with upper extremity DVT were less likely to have PE at presentation (9% compared to 29% for lower extremity DVT).12 However, during 90-day follow-up, patients with upper extremity DVT developed new PE at the same rate as those with lower extremity,12 and additional studies report substantial risks of PE (25%), clot recurrence (8%), or death (24%) following upper extremity DVT.13,14 The potential for upper extremity thrombi in the ICU has obvious implications for diagnostic strategies, which have traditionally focused on detecting lower extremity thrombi, as well as for therapeutic strategy with respect to vena caval interruption.
PE occurs when thrombi detach and are carried through the great veins to the pulmonary circulation. Pulmonary vascular occlusion has important physiologic consequences that lead to the manifestations of illness as well as to clues to diagnosis. The most profound effects of PE are evident on gas exchange and the circulation.
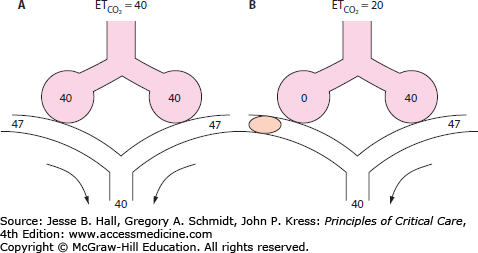
Gas Exchange: Physical obstruction to pulmonary artery (Pa) flow creates dead space in the segments served by the affected arteries. This creation of dead space has several effects on PCO2 and end-tidal CO2 (ETCO2), which can provide clues to diagnosis. If minute ventilation (VE) does not change, as occurs in a mechanically ventilated, muscle-relaxed patient, PCO2 will rise. However, most patients augment VE more than necessary to maintain elimination of CO2, so that PCO2 typically falls with PE. In health, the ETCO2 is nearly identical to arterial CO2. After pulmonary embolization, since end-tidal gas is a mixture of ventilated alveolar gas (in which PaCO2 approximates arterial, or PaCO2) as well as the newly created physiologic dead space gas (in which PaCO2 approximates inspired PCO2, or nearly zero), ETCO2 falls in proportion to the degree of dead space and no longer approximates PaCO2 (Fig. 39-1). This principle of a fixed alveolar to arterial gradient for PCO2 has been used to distinguish acute exacerbations of chronic obstructive pulmonary disease (COPD) from pulmonary embolism in patients with acute ventilatory failure.15 While not yet studied in the critically ill population, the steady-state end-tidal alveolar dead space fraction—which can be easily derived once one has both an accurate PaCO2 and PETCO2—has a sensitivity of 79.5% and a negative predictive value of 90.7% in hospitalized patients with PE.16 Similar studies report the utility of alveolar dead space fraction (VADS/VT) when used in conjunction with D-dimer for evaluating emergency department patients suspected of having PE.17 However, shortcomings to the VADS/VT approach include the technical challenge of simultaneously obtaining a steady-state exhaled gas PCO2 via volumetric capnography and PCO2 from an arterial gas sample.
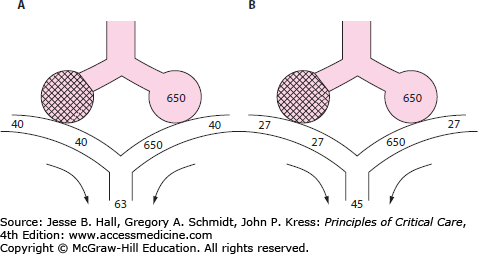
FIGURE 39-1
End-tidal CO2 in PE. Panel A demonstrates the normal end-tidal CO2, reflecting the alveolar CO2 of 40. Panel B illustrates the effect of obstruction of blood flow to half the ventilated alveoli. The end-tidal CO2 falls in proportion to the fraction of ventilated alveoli which are no longer perfused. All numbers are 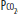 , in mm Hg.
, in mm Hg.
In 2010, a more simplified application of the same principle was tested by evaluating the combination of exhaled end-tidal CO2/O2 and D-dimer in emergency department, hospital ward, or ICU patients suspected of PE undergoing multidetector-row CTPA.18 As alveolar dead space fraction rises, the ratio of CO2/O2 falls. A CO2/O2 ratio <0.28 was considered positive for increased dead space.18 Among moderate-risk patients with a positive D-dimer, the presence of ETCO2/O2 <0.28 significantly increased the posterior probability of segmental or larger PE, and no segmental or larger clots were observed in patients with CO2/O2 >0.45.18 The alveolar dead space fraction estimated by this method is not sensitive to detecting PE at or below the subsegmental level, and the majority of measurements fall in an intermediate, and thus potentially clinically unhelpful, range. Furthermore, patients with hemodynamic instability or those already dosed with thrombolytic therapy were excluded in this study, and this study was not designed to test the safety of withholding further testing or anticoagulation based on D-dimer and CO2/O2 results. While we cannot advocate routine use of ETCO2/O2 in evaluating patients suspected of PE, an unexplained high dead space fraction in an ICU patient should prompt consideration of PE.
A widened alveolar to arterial gradient for oxygen (A-a)PO2 is present in the majority of patients with PE.19 However, since in PE hyperventilation is the rule, PaO2 may not be low. In unselected patients with PE, only 50% to 60% demonstrate a PaO2 <70 on ABG testing.19,20 Therefore, a normal PaO2 does not conclusively exclude a diagnosis of PE. There have been several efforts to use various combinations of the PaO2, the PCO2, and the (A-a)PO2 to predict the likelihood of pulmonary embolism, but these have all been shown to have insufficient discriminant value, even in patients without antecedent cardiopulmonary disease.21
Mechanisms of hypoxemia have been elucidated by applying the multiple inert gas elimination technique (MIGET) to patients with PE.22,23 Increased shunt fraction is found in only a few patients. In some, this may be due to opening of a probe patent foramen ovale when right atrial pressure rises following PE (discussed below), with a consequent intracardiac, right-to-left shunt. Atelectasis due to impaired surfactant production may also contribute to shunt physiology. In most patients, however, the most important contributor to hypoxemia is mismatching of ventilation and perfusion. In addition, the fall in cardiac output (˙Qт) that accompanies most pulmonary emboli leads to a fall in mixed venous saturation. This lowered venous saturation magnifies any hypoxemia due to shunt or ventilation perfusion (V/Q) mismatch (Fig. 39-2). Experimental studies suggest histamine release during acute PE may also lead to bronchoconstriction,24 worsening V/Q mismatch.
FIGURE 39-2
Effect of the mixed venous oxyhemoglobin saturation on arterial saturation in the setting of venous admixture. Panel A shows the effect of a 50% shunt on 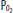 in a patient breathing 100% oxygen who has a normal mixed venous
in a patient breathing 100% oxygen who has a normal mixed venous  of 40 mm Hg (75% saturation). Panel B illustrates the same patient after the mixed venous
of 40 mm Hg (75% saturation). Panel B illustrates the same patient after the mixed venous 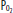 has fallen to 27 (50% saturation). Note that the
has fallen to 27 (50% saturation). Note that the 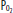 and saturation have fallen significantly despite the fact that the
and saturation have fallen significantly despite the fact that the 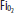 and the lungs have not changed at all. All numbers are
and the lungs have not changed at all. All numbers are 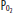 , in mm Hg.
, in mm Hg.
While impaired oxygenation is important and often provides a clue to the diagnosis of PE, the oxygen deficit is typically responsive to modest oxygen enrichment of inspired gas. Severe hypoxemia is usually seen only in patients with profound shock. If oxygen-refractory hypoxemia is present without obvious hypoperfusion in a pt with a PE, a patent foramen ovale should be suspected. Hypercapnic ventilatory failure is an uncommon presentation for patients with normal lung mechanics, since most patients can double or triple VE to maintain a normal (or reduced) PCO2. This compensatory response may be blunted in patients with preexisting lung disease, however. Typically, the more severe impact of PE is on the circulation, not on gas exchange. With some exceptions (eg, some patients with severe COPD), morbidity and mortality from PE relate to cardiovascular compromise, not respiratory failure.
Circulation: PE obstructs the pulmonary vascular bed both mechanically and via humoral mechanisms (thromboxane and histamine release).24,25 Pulmonary vascular obstruction increases right ventricular afterload which, compounded by tachycardia, increases right ventricular oxygen consumption. The right ventricle dilates and thins, its wall tension rises, and coronary perfusion is impeded.25 At the same time, pulmonary vascular obstruction compromises ˙Qт and contributes to hypoxemia. Therefore, just at the time when the right ventricle demands an increased oxygen delivery, the left ventricle may be unable to supply it. The superposition of increased right heart oxygen demand on decreased oxygen supply places the right ventricle at risk for ischemia, precipitating failure of the right heart (cor pulmonale). Acute right heart dysfunction, further discussed in Chap. 38, is the likely cause for sudden death in patients with massive PE (Fig. 39-3).
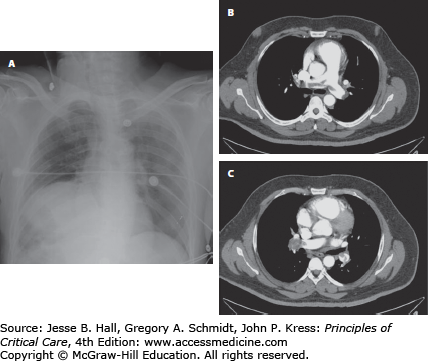
FIGURE 39-3
Radiographic signs sometimes observed in pulmonary embolism. Panel A: Westermark sign with oligemia of the right hemithorax relative to the right. VQ scan revealed an isolated perfusion defect of the right lung. Panels B and C are CT angiogram images revealing a saddle and right main pulmonary arterial embolus. Despite the massive appearance on CT, the patient was not in shock, and improved on heparin alone. A CT scan done 5 days later revealed clot only at the segmental level.
Of particular interest to the intensivist is the patient with a sublethal, yet large, PE. With increased afterload, the right ventricle dilates to a larger end-diastolic volume. This is associated with elevated right atrial and ventricular pressures and abnormally low ˙Qт. One consequence of raised right atrial pressure is the potential for right-to-left shunting across a probe patent foramen ovale, causing oxygen-refractory hypoxemia. Intracardiac shunting could in turn allow paradoxical embolization of thrombus to the systemic arteries, with resultant stroke or systemic occlusive symptoms. The increase in right ventricular pressure and volume also affects the left heart. A change in shape of the right ventricle and corresponding shift of the interventricular septum from right to left alters the diastolic pressure-volume characteristics of the left ventricle (Fig. 39-4) and may be detected echocardiographically. The resultant fall in left ventricular (LV) compliance impairs diastolic filling, reducing LV preload and further limiting ˙Qт. In experimental models, reduced LV preload appears dependent on increased pericardial constraint as the pulmonary vascular resistance rises.26,27 Increased pericardial constraint may explain why pulmonary artery occlusion pressure, meant to approximate LV end-diastolic pressure, typically does not fall during PE, and may be a poor indicator of LV preload.26
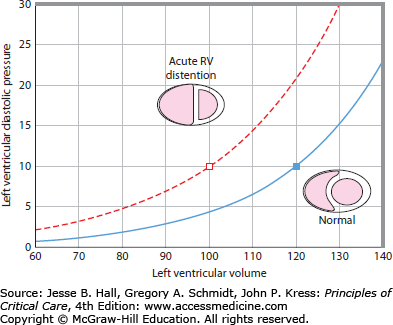
FIGURE 39-4
Left ventricular diastolic pressure-volume (PV) relationship before and after right ventricular dilation. The normal PV relationship (solid line) shows that large increments in diastolic volume are accompanied by small changes in pressure. With right ventricular distention (broken line), increments in volume are associated with relatively greater pressure changes. Note that at the same LV filling pressure of 10 mm Hg, the normal ventricle contains 20 mL more blood than following RV distention. This is associated with a correspondingly higher stroke volume and  . Pressures are in mm Hg, and volumes are in mL.
. Pressures are in mm Hg, and volumes are in mL.
CLINICAL MANIFESTATIONS
Most patients with PE will complain of dyspnea, chest pain, and apprehension.19,28 Less common symptoms are cough, diaphoresis, and hemoptysis. Up to 42% of subjects with PE will complain of leg or thigh symptoms, though in the PIOPED II registry, the same complaints were noted in 20% of subjects who did not have PE.19 Syncope is uncommon but described in all large series of PE (Table 39-1).
The majority of patients will demonstrate tachypnea and tachycardia. Pleural rub and signs of DVT are seen only occasionally. Fever is more common than is generally appreciated and seen in half the patients,28 but only rarely is the temperature greater than 38.5°C. Patients with large emboli may have the typical findings of any patient with low output shock such as hypotension, narrow pulse pressure, and poor peripheral perfusion. Occasionally, unanticipated failure to come off mechanical ventilation or unexplained episodes of respiratory distress may be hints to a diagnosis of PE. Rare patients with PE have disseminated intravascular coagulation, systemic embolization, or ARDS as their presenting manifestation.
Most patients will demonstrate hypoxemia or at least a widened (A-a)PO2. However, since a small but significant fraction will have normal oxygenation,20 the blood-gas value should not dissuade a physician from considering the diagnosis when the rest of the clinical picture is suggestive. As described above, the blood gas does not provide data which is useful in discriminating patients with PE from those without. Plain chest radiography can be helpful, in that only 12% of patients with PE have a normal film.29 The chest x-ray may demonstrate areas of oligemia (Westermark sign) and rare patients will develop a pleural based, truncated cone (Hampton hump); however, these “pathognomonic” signs are poorly sensitive, and most films have only nonspecific findings.29 In fact, the greatest utility of the chest film is in making alternative diagnoses such as pneumonia, pneumothorax, or aortic dissection. Nevertheless, the typical (albeit nonspecific) findings of basilar atelectasis, elevation of the diaphragm, and pleural effusion should always suggest PE when there is no ready alternative explanation. Electrocardiography (ECG) may reveal signs of right heart strain such as rightward axis shift, right bundle branch block, or right precordial strain but often shows only sinus tachycardia. PE has also been described to cause coved ST elevation and Q waves anteriorly in the setting of a normal cardiac angiogram, which resolved with anticoagulation.30
Valuable signs of PE may come from many of the devices used to monitor critically ill patients. The intensivist may derive clues from the ventilator, expired gas analysis, the Pa catheter, or during echocardiography. The sensitivity and specificity of these monitors for the diagnosis of PE are not known. Nevertheless, by incorporating such data, clinicians may alter their PE risk assessment and improve the benefit to risk ratio for subsequent testing and therapy.
The Ventilator: To maintain PCO2, the patient with PE must augment minute ventilation. Therefore, any unexplained increase in VE should prompt consideration of PE. Of course, any cause of rising dead space (airflow obstruction, hypovolemia, PEEP) or increased CO2 production (anxiety, pain, fever, sepsis) will also increase VE. However, when none of these conditions is apparent, especially when supporting clues are evident, PE becomes more likely.
Expired Carbon Dioxide: As described above, the increment in dead space after PE causes a detectable fall in ETCO2. With technologic improvements in these devices, noninvasive assessment of expired CO2 is becoming increasingly practical in the ICU. A corollary of the fall in ETCO2 with PE is that if VE does not rise (eg, in a muscle-relaxed or highly sedated patient), the total excretion of CO2 (expired CO2 concentration × VE) must fall. Therefore, PaCO2 will rise progressively until a new steady state is reached at a higher PaCO2. This can be demonstrated numerically by the alveolar dead-space fraction (AVDSf) as follows:
When combined with a negative D-dimer value, as will be discussed shortly, an AVDSf of less than 0.15 has been shown to exclude PE in hospitalized patients with a sensitivity of 97.8% and a negative predictive value of 98%.16 In calculating the AVDSf, however, one must take care to ensure a properly calibrated blood gas analyzer, as even small changes in PaCO2 measurements will cause large differences in AVDSf. Strong consideration of PE is warranted whenever a rising arterial PCO2 is noted in the setting of relatively constant CO2 production.
Pulmonary Artery Catheter: The most obvious clues from the pulmonary artery catheter (PAC) are the elevations in right atrial, right ventricular, and Pa pressures and concomitant fall in ˙Qт that occur with PE. Concomitant with reduced ˙Qт, one observes widening of the arterial (A) to venous (V) oxygen content difference (Fick principle) and a decrement in the mixed venous oxygen saturation (SvO2) or the central venous oxygen saturation. A final clue from the PAC may lie in the difference between the Pa diastolic pressure and the pulmonary artery occlusion pressure (Paop), though rising pericardial constraint may blunt a fall in Paop.26 Normally, flow through the pulmonary circulation is pulsatile, so that by the end of diastole, there is no more flow from the Pa to left atrium. Without flow, there can be no pressure gradient from the Pa to left atrium. Thus the end-diastolic Pa pressure and the Paop are nearly equal. When there is obstruction of the pulmonary vascular bed, however, flow is not completed by the end of diastole and a pressure gradient remains. A discrepancy between the Pa diastolic pressure and Paop may provide a clue to Pa obstruction.31
Unfortunately, such pulmonary arterial changes are both nonspecific and insensitive, so that only rarely do such changes indicate PE. For example, cardiac dysfunction (systolic or diastolic) causes a rise in right heart pressures and a fall in Q˙т, any cause of low Q˙т will result in a widened A-V oxygen content difference; and any cause of acute lung injury or global hypoxic vasoconstriction may raise the Pa diastolic to Paop gradient. A further layer of complexity is added by observations that in randomized trials, PAC use results in a small but significant increase in the risk of PE compared to central venous catheters.32,33 Given the limitation of the PAC as a diagnostic tool and the risk of actually causing PE, it cannot be advocated for the diagnosis of PE.
Echocardiography: Intensivist-performed, goal-directed echocardiography occasionally points to PE as the cause of cardiopulmonary failure (Chap. 29). Similarly, a formal study requested for evaluation of a low flow state may unexpectedly reveal findings strongly suggestive of PE.34 These include a dilated, thin-walled, poorly contracting right ventricle, bowing of the interventricular septum to the left, or McConnell’s sign. Very rarely, echocardiography may demonstrate a thrombus in the right atrium or right ventricle (Table 39-2), clinching the diagnosis of PE. While its attractions include portability—especially for the evaluation of critically ill patients—non-invasiveness, potential to elucidate competing diagnoses (such as myocardial infarction or pericardial disease), and rapid availability, echocardiography is insensitive, and should not be used to exclude PE. In prospective trials of unselected patients, sensitivities of between 29% and 52% are reported for various echocardiographic criteria of right ventricular strain or dysfunction or tricuspid regurgitation.35,36 Many patients with PE have normal echocardiograms.
Echocardiographic Signs of Pulmonary Embolism
|
When echocardiography exhibits right ventricular dysfunction, it reliably predicts an increased risk of mortality from pulmonary embolism. One study examined 126 patients with PE with echocardiography on the day of diagnosis, and found moderate RV dysfunction to impart a sixfold increased risk of in-hospital death compared to normal RV function.37 Even in patients assessed to be hemodynamically stable at presentation, right ventricular dysfunction portends a worse prognosis; one study found that 10% of such patients develop shock and 5% died in the hospital, compared to a 0% mortality amongst patients with normal RV function.38 In another series of hemodynamically stable patients, recurrent embolism was strongly associated with baseline echocardiographic abnormalities in right ventricular wall motion.39 A word of caution is prudent, however, in that the classic echocardiographic findings of PE are nonspecific, being common to a number of causes of acute right ventricular pressure overload such as the acute respiratory distress syndrome, other forms of severe hypoxemia, or status asthmaticus (see Chaps. 52 and 55).
DIAGNOSIS
The typical critically ill patient is unable to complain of the usual symptoms of PE, has numerous explanations for tachycardia and tachypnea, is hemodynamically unstable, and is a poor candidate for transport for radiographic studies. For that reason, it is important to have a clear sense of the probability of PE in any given patient. Such a judgment is complex, and validated algorithms for determining prior probability in critically ill patients are not available. The clinician must synthesize the patient’s risk factors and cardiopulmonary physiology to arrive at a risk determination. In the following sections, the contribution of various tests in evaluating suspected PE is discussed. An approach to diagnosis is summarized in Figure 39-5.
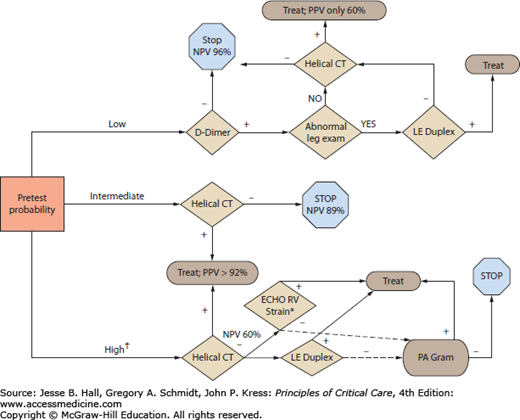
FIGURE 39-5
Diagnostic algorithm for critically ill patients suspected of having a pulmonary embolus. Interpretation of diagnostic tests is strongly dependent on the prior probability, or pretest clinical suspicion of PE. In patients with high clinical probability of PE, it is appropriate to empirically initiate heparin while pursuing the diagnostic work-up (†). The D-dimer test is recommended only for patients deemed to be low risk for PE; if negative in these patients, it is safe to withhold anticoagulation. Lower extremity duplex is the test of choice in all patients with leg symptoms, and it is highly sensitive in this setting and avoids the use of contrast; however, the negative predictive value (NPV) of the duplex is inadequate to rule out PE in intermediate or high risk patients. Based on PIOPED II,42 the positive (PPV) and negative predictive values (NPV) are shown for CT angiograms in different clinical settings. Note that when the CT result is discordant with the clinical probability, the test characteristics falter. Thus we recommend further testing for PE in patients deemed to be high risk for PE but in whom the CTA is negative; in this population, the NPV is only 60% and cases of PE could be missed. In such a case we advocate consideration of echocardiography and if right ventricular strain or dysfunction is discovered without alternative cause (*)—such as profound hypoxemia causing hypoxic vasoconstriction, or known pulmonary hypertension with evidence of chronic RV overload—we advocate treating with heparin or pursuing pulmonary angiography.
Since the symptoms, signs, and laboratory findings of PE are usually nonspecific, to wait for a patient with classic, unmistakable clues before pursuing a diagnosis risks missing the majority of patients with this potentially lethal disease. However, since nonspecific indicators of potential PE are ubiquitous, indiscriminant pursuit of the diagnosis is prohibitively costly and dangerous. Most patients with PE have identifiable risk factors (Table 39-3). Absence of risk factors for VTE should lead the physician to seek alternative explanations for the patient’s findings. On the other hand, when numerous risk factors are present, the diagnosis should be more seriously considered.
Risk Factors for Pulmonary Embolism
| Epidemiologic Factors | Obesity |
|---|---|
| Prior VTE | |
| Age | |
| Cigarette smoking | |
| Malignancy, especially adenocarcinoma histology | |
| Chronic obstructive pulmonary disease | |
| Pregnancy and postpartum | |
| Nephrotic syndrome | |
| Chemotherapy | |
| Estrogen therapy | |
| Venous stasis factors | Immobility |
| Paralysis | |
| Leg casts | |
| Congestive heart failure | |
| Pregnancy | |
| Varicose veins | |
| Prolonged travel | |
| Indwelling catheters (central or pulmonary arterial) | |
| Endothelial injury factors | Surgery |
| Trauma | |
| Postpartum state | |
| Hypercoagulability | Factor V Leiden polymorphism |
| Protein C or S deficiency | |
| Antithrombin-III deficiency | |
| Activated protein C resistance | |
| Antiphospholipid antibody syndromes | |
| Polycythemia | |
| Macroglobulinemia | |
| Thrombocytosis | |
| Heparin-induced thrombosis |
Given the nonspecific presentation of most patients with PE, a clinical risk prediction tool has been developed to help stratify patients with a possible diagnosis of PE and to identify a low-risk group of patients for whom further testing is unnecessary. Known as the Wells criteria,40 the most often cited prediction rule is shown in Table 39-4, and for stable patients, it appears safe to withhold anticoagulation when the score is ≤4.0 and the D-dimer test is negative. The Wells criteria have not been tested in critically ill populations, but the assessment of a patient’s global risk for PE based on patient historical factors, clinical presentation, and differential diagnosis remains a necessary step. Because PE lacks a perfect diagnostic test, the clinician must synthesize both pretest probability and test results in order to select the most rational therapy for each patient.
Wells Clinical Decision Rule for PE
| Clinical Factor | Points Assigned |
|---|---|
| Clinical signs or symptoms of DVT present | 3.0 |
| Alternative diagnosis is LESS likely than PE | 3.0 |
| Heart rate >100 beats/min | 1.5 |
| Immobilization >3 days OR surgery within prior 4 weeks | 1.5 |
| Previous VTE | 1.5 |
| Hemoptysis | 1.0 |
| Malignancy | 1.0 |
The gold standard for the diagnosis of PE has long been the pulmonary angiogram. Its use in critically ill patients is limited, however, by invasiveness, expense, need for dye infusion, and the risks attendant to transport out of the ICU. In many patients, the pulmonary angiogram can be replaced by its noninvasive cousin, the multidetector row helical computed tomography pulmonary angiogram (CTPA). CT angiography has supplanted both the ventilation perfusion (V/Q) lung scan and pulmonary angiography in the diagnosis of PE, and is currently the recommended modality of choice for patients with a moderate or higher pretest probability of PE.41,42 In this section we will review both traditional and modern radiographic testing for PE, and consider the added benefit of ancillary tests such as biomarkers and noninvasive leg studies. An integrated approach to the diagnosis of PE is described in Figure 39-6.
FIGURE 39-6
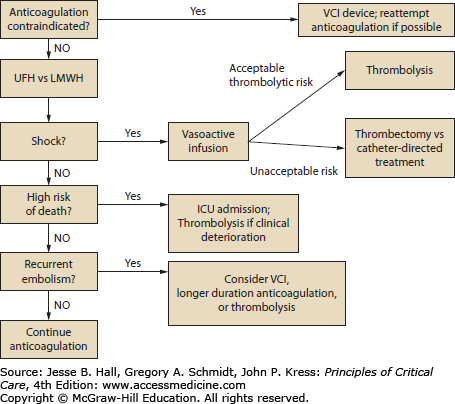
Treatment of PE in critically ill patients. Once a PE is diagnosed, the treatment of choice is anticoagulation. For patients in shock, thrombolytics are the preferred treatment unless the risk of these agents is deemed unacceptably high; surgical or catheter-directed treatment may be considered in that situation, though both are more effective with subsequent anticoagulation. For submassive PE or those patients deemed to be high risk for progressing to shock or death, ICU admission and close monitoring is recommended, with escalation of therapy if there are signs of clinical deterioration.
Computed Tomography (CT) and Magnetic Resonance Imaging (MRI): Technical advances in imaging modalities have made computed tomography pulmonary angiography (CTPA) and gadolinium-enhanced magnetic resonance angiography (MRA) very attractive diagnostic modalities for PE, not least due to the trust we place in images. With these modalities, the clinician can “see” the filling defect representing clot. Furthermore, both CTPA and MRA may diagnose alternative conditions to explain the patient’s symptoms. Both are less invasive and less expensive than pulmonary angiography, and CTPA particularly is faster and easier than V/Q scanning to perform in a critically ill patient.
Multidetector-row CTPA produces a two-dimensional image of the lung and its vessels at very small collimeter, or slice thickness. It has been shown to detect central emboli—out to fourth-division vessels—with a high degree of reliability. While CTPA protocol is minimally invasive, it does require the injection of intravenous contrast, is expensive—though less so than either MR or pulmonary angiography—to perform and to interpret, and can be difficult to perform in patients who are unable to hold their breath or who are hemodynamically unstable. The biggest critiques of CTPA have been its questionable ability to detect emboli to the level of subsegmental artery, and interobserver variation in CTPA interpretation. However, the technology of helical CT has grown exponentially in the past 15 years, from a single detector collecting data at fixed intervals as it rotates around the patient during a single breath hold, to much faster rotation and multiple channels simultaneously collecting data. Whereas a single-row detector captured approximately 1 slice per second, a 16-row scanner rotating faster can acquire 40 slices per second.43 Combined with thinner collimation, the result is faster image acquisition, decreased motion artifact, and higher-resolution images.
To answer these critiques in the era of modern multirow detector scanners, which vary from 4- to 128-row and beyond, the multicenter prospective investigation of PE diagnosis II (PIOPED II) study was undertaken. This trial performed CTPA and CT venography of the IVC and lower extremities in 842 subjects referred for suspected PE.42 All subjects underwent a risk assessment prior to their diagnostic testing (Wells criteria Table 39-4),40 and sensitivity and specificity of CT were considered in comparison to composite reference standards for both the presence and absence of PE. A positive PE diagnosis was considered if the subject had a high probability VQ scan; an abnormal digital subtraction pulmonary angiogram (DSA); or the combination of an abnormal lower extremity venous ultrasound with a nondiagnostic VQ scan (not high probability and not normal). Exclusion of PE by reference standard could occur by normal DSA; normal VQ scan; or low probability VQ scan, normal venous ultrasonography, and clinical Wells score <2.42 Approximately 7% of subjects had an uninterpretable CT scan. Of those with an interpretable study, the sensitivity and specificity of CTPA were 83% and 96%, respectively, giving positive and negative likelihood ratios of 19.6 and 0.18.42 These are characteristics of a very useful test, and most experts now recommend CTPA as the primary imaging diagnostic modality for patients suspected of PE with an intermediate or higher clinical risk assessment for PE.44
The elegant design of the PIOPED II trial highlights two important considerations in PE diagnosis, however. The first is the critical importance of performing a pretest risk assessment for PE, in order to interpret diagnostic results. When compared to the composite reference standard, CTPA (and combined CTPA-CTV, which increases sensitivity slightly without altering specificity) performs extremely well when the test result is concordant with the pretest probability. For example, in a patient deemed to have a high pretest probability for PE, the positive predictive value of CTPA was 96%. Similarly for a patient with low pretest probability, the negative predictive value was 96%. However, for subjects with discordant test results relative to their pretest risk assessment, the predictive values drop to only 60% (Table 39-5).42 Thus for patients in whom the clinical risk assessment is low probability for PE, a positive D-dimer test on hospital admission or an unexpected finding of right ventricular strain on echocardiography would be useful to support the utility of CTPA. Conversely, if the clinical assessment for PE is high, a negative CTPA should not necessarily terminate consideration of PE. In this setting, additional venous ultrasonography (if CT venography was not performed) or digital subtraction angiography may be warranted.
The PIOPED II study also addressed the ability of CTPA to detect subsegmental PEs, as this has been a point of controversy with CTPA, and observations were that interobserver variation became more prevalent with smaller clots.45,46 It is interesting to recall that in the original PIOPED study in 1990, subsegmental PEs accounted for 6% of the PEs detected, and that the interobserver agreement for pulmonary angiograms among these small PEs was wider than that of larger PEs (66% for subsegmental PEs compared to 90% for segmental and 98% of lobar PEs).47 It would appear that subsegmental PEs are challenging to diagnose even by pulmonary angiography. Furthermore, the significance of these smaller filling defects is highly uncertain. In PIOPED II, the positive predictive value of a filling defect on CTPA relative to the composite PE diagnosis fell dramatically from 97% for a main or lobar artery, to 68% for a segmental artery, to just 25% for a subsegmental clot.42 Based on retrospective review of data, some experts recommend that anticoagulation may be safely withheld for patients with exclusively subsegmental PE when certain conditions are met. In the absence of a prospective investigation, however, these recommendations seem to have limited applicability to the critically ill population, given that they require the patient to have adequate cardiopulmonary reserve and a transient, resolved risk factor for PE.48 The optimal treatment for subsegmental or smaller clots in the ICU remains uncertain.
Magnetic resonance angiography (MRA) technology has also progressed greatly in the past 2 decades, with rapidly improving resolution and speed of image acquisition. MRA does not require iodinated contrast, and does not have the risk of radiation associated with CTPA. Among initial reports in 1997, one study listed a sensitivity of 100% and specificity of 95% when MRA was performed in 30 patients undergoing pulmonary angiography for suspected PE49; another early study reported 77% sensitivity but 98% specificity.50 The initial optimism over MRI as a diagnostic tool for PE has been tempered by issues of obtaining sufficient quality scans in critically ill subjects. In the recent PIOPED III study, 25% of MRI studies were technically inadequate, and the sensitivity of MRA was under 80% even when technically adequate. When tandem pulmonary MRA and lower extremity MR venography were combined, both sensitivity and specificity were >97%, but over half of studies were technically inadequate.51 Thus the use of pulmonary MRA in diagnosing PE should be limited to centers with pulmonary MRA expertise and only for patients with a contraindication to more conventional testing.
Noninvasive Leg Studies: Because the majority of subjects with PE have detectable concomitant DVT, and up to 50% of DVT patients will have PE, the diagnostic strategy for VTE must include both entities.25 Noninvasive leg studies have traditionally included impedance plethysmography, phleborheography, venous Dopplers, and B-mode ultrasound scanning of leg veins, and now extend to CT or MR venography done in conjunction with pulmonary angiography. The technical details of these procedures and differences between them are beyond the scope of this chapter, and in practice, most centers now rely upon B-mode venous ultrasonography. Venous ultrasonography is extremely helpful in assessing a patient with symptomatic proximal deep venous thrombosis; in this population, the test demonstrates a sensitivity of 97%, positive predictive value of 100%, and negative predictive value of 100%.52 Unfortunately, ultrasonography performs less well in asymptomatic patients. Patients with symptomatic DVT are far more likely to have a proximal than a distal, or isolated calf vein, DVT.53 In contrast, most asymptomatic DVT are distal, and since fewer than half of patients with PE have leg symptoms,19 it seems that PE more commonly follows asymptomatic rather than symptomatic DVT.47 Thus a negative venous duplex should not exclude the diagnosis of PE in a patient with intermediate or high clinical risk assessment. Conversely, demonstration of DVT provides rationale for anticoagulation, and is accepted as evidence for PE if clinical suspicion is present.42 When anticoagulation was withheld on the basis of a negative venous duplex for noncritically ill patients, the rate of subsequent DVT within 6 months was low, approximately 2%, and mortality from PE was less than 0.1%.52 Unfortunately, one suspects that negative duplex results are less meaningful for critically ill patients, who retain significant risk factors for developing new DVT as long as they are in the ICU.
With increasing use of peripherally inserted central catheters and indwelling transvenous pacemakers, deep vein thromboses in the upper extremities are becoming increasingly common. Prospective registries for VTE have reported that between 5% and 10% of observed DVTs occur in the upper extremities,12,54 and this proportion is likely to be higher among critically ill patients, since this population is enriched for malignancy, central vein catheters, and cardiac devices. Venous ultrasound appears to remain both sensitive and specific (both ∼90%) in the setting of upper extremity DVT,55 despite the fact that the proximal subclavian and brachiocephalic veins cannot be directly visualized due to bony structures. The addition of color Doppler may reveal an abnormal flow pattern to suggest proximal DVT even when distal veins are patent and compressible, though Doppler has not been proven to improve sensitivity.55 In critically ill patients, we advocate the extension of venous ultrasound to all four extremities when evaluating for PE in a patient with risk factors for upper extremity clots, including malignancy, indwelling central catheter or cardiac device, prolonged critical illness, hypercoagulable state, or upper extremity trauma. Digital subtraction venography, using intravenous contrast, is an option when venous ultrasound and CTPA are nondiagnostic, but carries much of the risk of conventional angiography with respect to dye load and radiation exposure.
Ventilation Perfusion (VQ) Lung Scan: V/Q scanning was traditionally the initial test of choice in the evaluation of PE, but it has been largely supplanted by CT scanning given that a definitive positive or negative result is generally achievable in only ∼20% of critically ill patients.56 V/Q scans can be extremely helpful to the clinician when they provide either a high probability result—with an attendant specificity of 85%, ruling in the diagnosis—or a normal result, when the diagnosis of PE is virtually excluded.47
Related posts:






Full access? Get Clinical Tree


