KEY POINTS
To gain an understanding of the mechanisms and anticipatory management of brain tissue displacement (herniation) and intracranial hypertension.
To understand available brain monitoring devices in measuring ICP and to appreciate their role in guiding early interventions to avoid secondary brain injury as hesitation to monitor intracranial pressure dynamics, and to aggressively pursue ICP management likely accounts for the vast majority of secondary brain injury in patients with reduced level of consciousness.
To foster an individualized patient approach in addressing abnormal ICP and flow dynamics within the practice of neurocritical care. Understanding the indications for brain monitoring via real-time parenchymal blood flow, oxygen tension, and chemistry surveillance, as well as mastering the current recommendations in aggressive management approaches toward elevated ICP such as induced hypothermia, suppression of abnormal electrical discharges, and early surgical decompression are necessary tools for the neurocritical care clinician.
CONSIDERATION OF CEREBRAL PRESSURE AND FLOW DYNAMICS
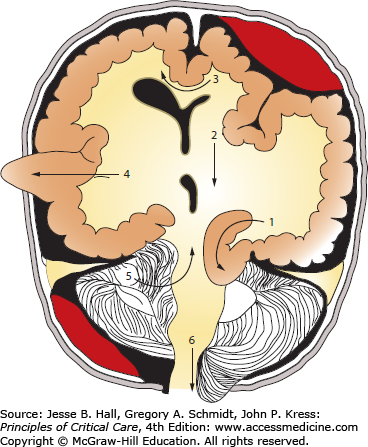
In adults, the cranial vault represents a closed, noncompliant structure. Two important exceptions exist in which intracranial compliance is increased. These are at the foramen magnum and craniectomy sites. Craniectomy refers to surgical bone removal to treat refractory intracranial hypertension or as a by-product of neurosurgical decompression for an alternate indication. This removal of bone leaves a palpable, soft, cranial defect covered only by dura, galea, and skin. The brain is distinguished from other organs by the unique challenge of monitoring brain function and intracranial dynamics in a structure enclosed by a bony vault. The noncompliant surrounding bone of the calvarium does not allow for significant volume change of the brain or adjustment of intracranial pressure (ICP) (Fig. 86-1A). As a result, the pressure within the fixed space of the calvarium must be carefully regulated by many mechanisms in order to be maintained within a physiologic range. Disruption of these mechanisms through trauma, space-occupying lesions, or edema leads to dysregulation of the delicate balance required to maintain normal pressure that results in significant neurologic and systemic dysfunction. For instance, the tentorial opening, which separates the supratentorial and infratentorial compartments, encloses, among other structures, the midbrain, posterior cerebral arteries, posterior communicating arteries, oculomotor, and sixth cranial nerves. These structures are frequently damaged during transtentorial herniation, leading to a chain of often irreversible, secondary injuries (Fig. 86-1B).
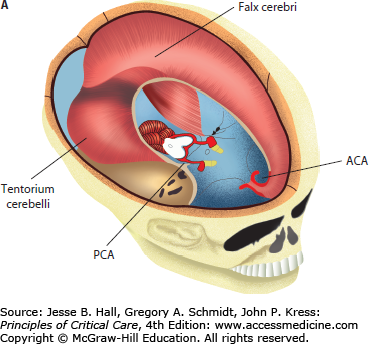
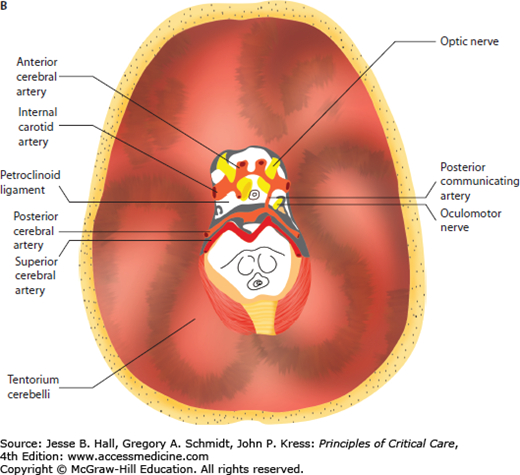
FIGURE 86-1
A. Anatomical relationship of key intracranial structures. The two hemispheres within the supratentorial compartment are separated and stabilized by rigid dura duplications, known as the falx and the tentorium, respectively. These structures become clinically important in the setting of brain herniations; for example, as a late complication of subfalcine herniation the anterior cerebral artery (ACA) is compressed against the free edge of the falx, leading to ACA infarction. Whereas in lateral or descending transtentorial herniation, the posterior cerebral artery (PCA) is displaced inferiorly over the free edge of the tentorium, leading to herniation-induced occipital lobe infarction. B. Tentorial opening and its contents. The tentorial opening that separated the supratentorial from the infratentorial space consists of the midbrain and important structures, that is, circle of Willis and cranial nerves. Due to the location of the oculomotor nerve, it is the most commonly affected nerve secondary to herniation of the medial temporal lobe and aneurysm of the posterior communicating artery.
The structures between the brain surface and the inner skull, the meningeal layers, are important in identifying and maintaining normal ICP. The most important of these is the subarachnoid space where the arachnoid villi conduct cerebrospinal fluid (CSF) from the subarachnoid space to the venous sinuses. If these granulations are blocked by inflammatory substances or disintegrated blood, nonobstructive hydrocephalus and increased ICP can result due to impaired CSF reabsorption. Other meningeal components are the subdural and epidural spaces where bleeding may occur, requiring immediate attention due to potential space-occupying lesions between these layers (Fig. 86-2).
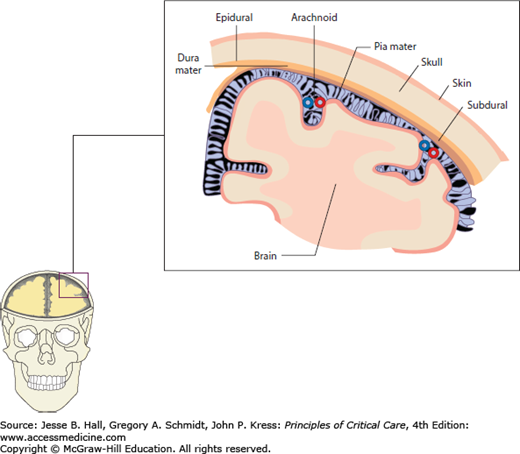
FIGURE 86-2
Meningeal layers. The cerebrospinal fluid compartments located between the brain surface and inner skull are clinically described as subarachnoid, subdural, and (functionally nonexisting) epidural spaces. Arachnoid villi within the subarachnoid space have the important role of continuous CSF absorption and, if obstructed due to infection or disintegrated blood products, communicating hydrocephalus and eventually elevated intracranial pressure will occur. The subdural and epidural spaces are important in that blood and fluid may track into and expand these potential spaces in the setting of trauma or ruptured vascular malformations.
The average volume within the cranium is 1500 mL, with ∼88% consisting of brain parenchyma, ∼7.5% composed of intracranial blood, and ∼4.5% composed of CSF.1 The sum of the partial pressures and volumes from these three main components is equal to the total ICP. Therefore, when one volume increases (eg, intraparenchymal brain tumor), the other volumes compensate for the pressure change and reduce their combined intracranial volumes to keep ICP constant. This is known as the Monro-Kellie doctrine. Frequent mechanisms responsible for maintaining a normal ICP (ie, <20 mm Hg) are a compensatory increase in CSF absorption, drainage of blood from the cerebral venous systems, and a shift of CSF from the cranial subarachnoid space into the spinal (intraforaminal) compartment. Because of skull noncompliance, any uncompensated changes in the volume have a significant impact on ICP and brain function. If untreated, sustained elevations in ICP may lead to compression of critical structures, vascular compromise with impaired cerebral perfusion, irreversible ischemia, and brain death.
The first ICP measurements were performed by Guillaume and Janny in 1951, but it was the seminal work of Nils Lundberg in 1960 who established intraventricular ICP monitoring using bedside strain gauge manometers to describe three ICP waveform patterns (A, B, C) associated with intracranial pathology. Of particular importance, the A-wave (or plateau wave) is observed with ICP increases between 25 and 75 mm Hg persisting for up to 20 to 25 minutes if left untreated. The rationale behind the study of ICP waveform and amplitude is that with each heartbeat there is a pulsatile increase in cerebral blood volume, the equivalent of a small intracranial volume injection, and the amplitude of the ICP pulse waveform is the response of ICP to that increment of volume. The properties of ICP wave should therefore be directly related to the craniospinal elastance.2
There is no level I evidence to support the use of a single ICP threshold to initiate therapy. The recommended critical ICP elevation in adults at which treatment should be initiated (Level II evidence) is 20 mm Hg sustained for more than 5 minutes.3 Failure of compensatory brain mechanisms to reduce ICP to normal values will result in intracranial hypertension and its clinical sequelae. Common etiologies for primary and secondary ICP elevations are listed in Table 86-1. Numerous studies have demonstrated that elevated ICP is associated with poor outcome and, therefore, that ICP control and pressure monitoring are among the key approaches to successful management of brain-injured patients.4-6 Too often, a general “cookbook” ICP management approach, without an understanding of the natural progression of the underlying injury and without real-time measurements of ICP, is applied leading to secondary brain injuries, which can exceed the magnitude of primary injury. A primary focus of neurocritical care, therefore, is to minimize secondary injuries. Examples of cases requiring continuous ICP monitoring are patients with large ischemic strokes and associated evolving edema; severe meningoencephalitis with generalized edema and hydrocephalus; and intracranial hematomas exerting local mass effect. These patients may require prolonged ICP monitoring in order to detect delayed cerebral edema or worsening primary injury. Another example of patients in need of invasive ICP monitoring are traumatic injuries, which may exhibit an otherwise undetected bimodal pattern of ICP elevations, or patients suffering from subarachnoid hemorrhage (SAH) who may develop ICP elevations due to undetected obstructive hydrocephalus or vasogenic edema secondary to vasospasm-induced ischemia.
Causes of Elevated Intracranial Pressure
| Primary (Intracranial) | Secondary (Extracranial) |
|---|---|
|
|
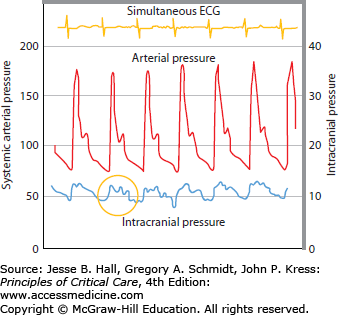
When ICP is monitored continuously, the tracing has a ballistic waveform similar to systemic arterial pressure (Fig. 86-3). The “pulse pressure” of ICP, however, is much narrower and is expressed, by convention, as a mean. The normal mean ICP is generally below 15 mm Hg, with an upper range at about 20 mm Hg, but its value will fluctuate in normal individuals depending on many physiologic factors such as head positioning, Valsalva maneuver, breathing pattern, etc (see Table 86-2).
Factors That Influence Cerebral Blood Flow and Intracranial Pressure
| Factor | Cerebral Blood Flow | Intracranial Pressure | Effect | Clinical Commentary |
|---|---|---|---|---|
| Raised intracranial pressure | Decrease | NA | – | Cerebral injury occurs through ischemia and mechanical compression |
| Cerebral hyperemia | NA | Increase | – | May be regional |
| Hyperventilation | Decrease | Decrease | Vasoconstriction | Prolonged hyperventilation leads to ischemia |
| Hypoventilation | Increase | Increase | Vasodilatation | Seen with posterior fossa pathology |
| Hypotension | ± | Increase | Vasodilatation | Early diagnosis and treatment is imperative |
| Hypovolemia | ± | Increase | Vasodilatation | Maintain euvolemia |
| Acidosis | Increase | Increase | Vasodilatation | Important in ICP control |
| Alkalosis | Decrease | Decrease | Vasoconstriction | Avoid in cerebral vasospasm |
| Hyperthermia | Increase | Increase | Vasodilation | Linear increase in cerebral blood flow 6% per °C |
| Hypothermia | Decrease | Decrease | Vasoconstriction | Therapeutic value |
| Hypoxia | Increase | Increase | Vasodilatation | Significant at <SPAN role=presentation tabIndex=0 id=MathJax-Element-1-Frame class=MathJax_Error style="POSITION: relative" data-mathml='PaO2′>[Math Processing Error]PaO2 <50 mm Hg |
| Increased intrathoracic pressure | Decrease | Increase | Cerebral venous outflow attenuation | Valsalva maneuver |
| Pain/arousal | Increase | Increase | Vasodilatation | Avoid noxious stimuli |
| Volatile anesthetics | Increase | Increase | Vasodilatation | Additive ICP increases with head down positioning during anesthesia |
| Seizures | Increase | Increase | Increased metabolism and Valsalva | Maintain low threshold for prophylactic antiepileptic drugs in ICP susceptible patients |
| Positive end- expiratory pressure (PEEP) | Increase | Increase | Decrease in cerebral venous outflow | Variable effect on intracranial pressure (Caution: PEEP of >12) |
A schematic diagram delineating the tight relationship between ICP and intracranial volume is depicted in Figure 86-4. Intracranial compliance is the association between changes in intracranial volume and expected changes in pressure, while the reciprocal is defined as elastance—a change in pressure leading to a change in volume. Intracranial compliance, although not measured directly in absolute numbers, is an important and frequently utilized clinical concept that can be readily estimated in an ICP-monitored patient. Compliance describes the fact that a disease process that increases or displaces the volume of a component of the intracranial cavity will first be compensated for by a decrease in the least resistant compartment—the subarachnoid CSF spaces, which are contiguous over the convexities and within the cisterns and ventricles. As a result, ICP may increase only minimally while a reserve for intracranial compliance exists during the early stages of the disease process. The ICP waveform (but not its mean pressure) may already indicate a decline in brain compensatory mechanisms, however (Fig. 86-4B). Once CSF cannot be passively displaced any further, the ICP rises more sharply as the reserve for compliance decreases (Fig. 86-4C). At this point, blood vessels begin to provide an element of compliance, and will compensate for ICP increases by extruding blood out of the intracranial space. First, the less resistant cerebral venous system reduces its blood volume, and then the circulation within the arterial tree is reduced. Lastly, brain parenchyma will follow by shifting along the ICP gradient within the cranial vault and away from the space-occupying lesion. This is described as brain herniation. Herniation syndromes can be distinguished clinically and radiographically depending on which vector the ICP gradient continues to evolve, that is, from one to the opposite hemisphere or along the craniocaudal cerebrospinal axis.
FIGURE 86-4
Intracranial compliance. This curve represents the compliance function of the brain. With added volume, the intracranial compartment initially shows good compensatory reserve identified by normal ICP (A). Additional volume increases are still tolerated (ie, high-normal ICP readings) but further reducing the compensatory reserve (B; worsening compliance). At a critical point, compensation of intracranial compliance diminishes exponentially (C; exhausted compensatory reserve), leading to abrupt ICP elevations.
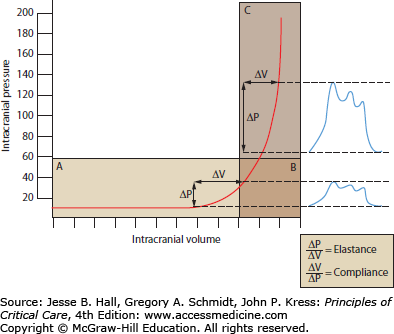
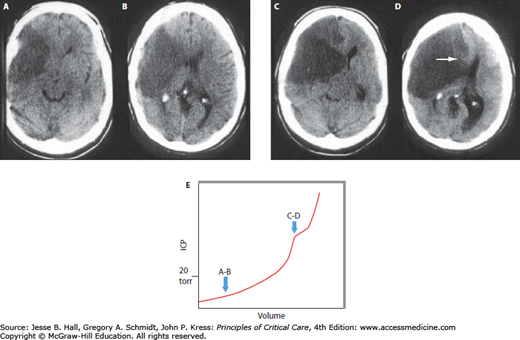
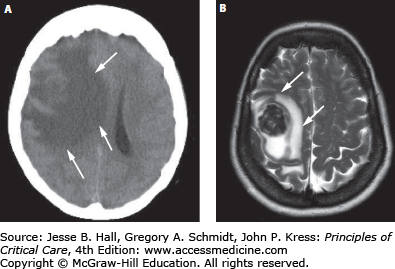
The important concepts of ICP-volume relationships and intracranial compliance can be applied to the radiographic estimation of the likelihood of intracranial hypertension from a space-occupying intracranial process to determine the indication for invasive pressure monitoring. For example, Figure 86-5A and B show a brain CT scan identifying vasogenic edema from a right middle cerebral artery (MCA) territory infarction. As there are still compressible spaces visible around the swollen brain (eg, basal cisterns, ipsilateral lateral ventricle, and ipsilateral cortical sulci), it is reasonable to expect that the ICP is not yet significantly or persistently elevated. In contrast, the CT in Figure 86-5C and D shows a more extensively swollen hemisphere than Figure 86-5A, with obliteration of all surrounding CSF spaces. The ICP is therefore predicted to be markedly elevated. While estimating the likelihood of intracranial hypertension by radiographic appearance is imperfect, it can provide some practically useful guidance in management decisions when the clinician is forced to initiate invasive ICP monitoring or to consider other therapeutic maneuvers such as surgical decompression.
FIGURE 86-5
Neuroimaging and intracranial compliance. Head imaging does not replace ICP monitoring; however, some estimates of intracranial compliance can be obtained. The head CT (A and B taken at 48 hours postevent) identifies right middle cerebral artery ischemic infarction with local mass effects; however, there are remaining compressible CSF spaces (ventricular system, basal cisterns) visible indicating reduced but not exhausted intracranial compliance. With further mass effects and tissue shift (C and D; CT taken at about 96 hours) almost complete compression of neighboring CSF spaces and exhausted intracranial compliance is identified in addition to evolving herniation-induced (right to left subfalcine herniation) led to new right anterior cerebral artery infarction (arrowhead in D). The estimated, relative intracranial compliance for the obtained head CT scans is delineated on the graph below (E).
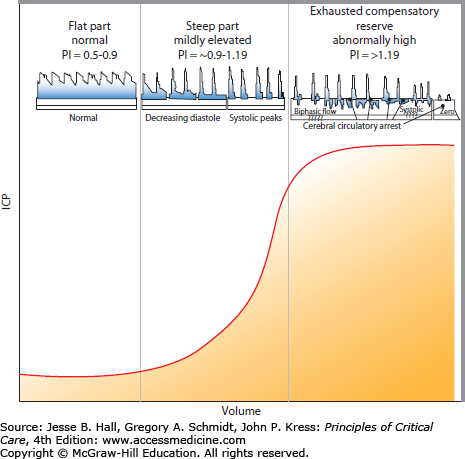
To appreciate the progressive detriment of elevated ICP, it is essential to understand the factors involved in determining and controlling cerebral blood flow (CBF). Neglecting, for a moment, that cerebral arteries are rather flexible conduits, CBF could be compared to electric current through a wire. Ohm described that this current (I) is proportional to the difference in the potential (ΔV) placed across the ends of a wire and proportionally constant to the resistance (R) the current faces while traveling through the wire. That is, current = potential difference/resistance (I = ΔV/R) or (ΔV = IR). Written in flow dynamic terms, CBF depends on the perfusion pressure (CPP) divided by the vascular resistance (CVR) or CBF = CPP/CVR. As CPP is calculated by the difference between the mean arterial pressure (MAP) and the ICP, this equation can be rewritten as CBF = MAP − ICP/CVR. CVR is governed by precapillary, brain penetrating arterioles and is tightly regulated by pressure autoregulation in a normal patient to provide a steady CBF with normal MAP fluctuations. Autoregulation is a function of vasoactive mediators between neighboring vascular endothelial cells, adjacent smooth muscle, and perivascular nerves.7,8 Dynamic increases in ICP can also be estimated at the bedside by elevated blood flow velocities and pulsatility indices as seen on transcranial Doppler (TCD). CVR changes can be exhausted, however, leading to complete absence of flow if increased ICP becomes intractable (Fig. 86-6).
FIGURE 86-6
Intracranial pressure-volume curve correlated with blood flow velocities. A relative relationship exists between intracranial compliance, intracranial pressure (ICP), cerebral perfusion pressure (CPP), and the pulsatility index (PI), which is obtained from the blood flow velocity profiles measured by transcranial Doppler (TCD). The normal PI value ranges from about 0.5 to 1.19 and correlates best with normal compliance and ICP ranges (identified on the left column). The PI starts to increase approximately starting from 0.9 to 1.19 with compromised intracranial compliance even when ICP still remain normal (middle column). The PI further increases from about >1.19 with exhausting intracranial compliance, ICP elevation, CPP reduction, and decreasing vascular bed, which eventually leads to circulatory arrest (right column). Characteristic TCD flow velocity waveform changes with increasing PI are represented above the graph. TCD is a helpful and readily available bedside technology to monitor intracranial compliance.
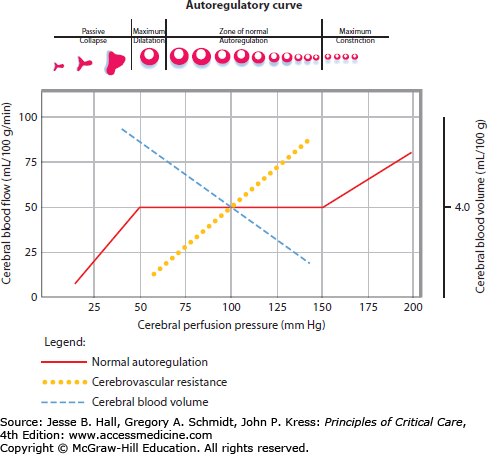
Alterations in CBF and CVR can disturb autoregulation homeostasis, leading to primary and secondary cerebral injury. A graphical depiction of CBF remaining constant over a wide range of arterial blood pressures, at least in the normotensive noninjured brain, is presented in Fig. 86-7. In chronically hypertensive individuals, the autoregulatory threshold is shifted to the right. Relative blood pressure lowering within the autoregulatory range will be compensated by cerebral vasodilation and a resultant increase in cerebral blood volume (CBV). Conversely, relative blood pressure elevations within an individual’s autoregulatory range leads to cerebral vasoconstriction and a subsequent decrease in cerebral blood volume. The physiologic relationship between blood pressure, CBF, CPP, and CVR is unpredictable in damaged brain regions with impaired autoregulation. Both ischemia (regionally due to arterial occlusion or globally as in ischemic encephalopathy following cardiac arrest) and CBV dysregulation (eg, hyperemia) are critical determinants of ICP, especially in the noncompliant, autoregulatory-paralyzed brain already exposed to elevated ICPs from the primary injury. This practical understanding of cerebral hemodynamics, and the concept that CBF in the injured brain is almost entirely dependent on MAP, is critical for the development of a rational therapeutic plan for patients with brain injury and intracranial hypertension.
FIGURE 86-7
Autoregulatory curve. With intact cerebral autoregulation the cerebral blood flow is maintained constant over a wide range of cerebral perfusion pressures (50-150 mm Hg; solid red line). Outside of this pressure range, cerebral arterioles collapse at very low CPP or at very high pressures abnormally (breakthrough) constricts. Abnormal autoregulation, commonly present in injured brain, is the complete dependence of cerebral perfusion on systemic arterial pressures, an abnormality that has important consequences on intracranial pressure.
Regional CBF normally averages 50 to 60 mL/100 g/min, about 15% of the cardiac output (about 700 mL/min). Assuming normal cellular metabolic rate, increased oxygen extraction from the blood compensates for reduced CBF until CBF reaches 50% of its baseline value, when the first clinical and electroencephalographic (EEG) manifestations of hypoperfusion appear. Impairment of cortical activity becomes more marked at 16 to 18 mL/100 g/min with loss of neurotransmission due to Na-K pump failure. Cytotoxic edema then occurs at 10 to 12 mL/100 g/min. At ranges of 6 to 10 mL/100 g/min, progression to calcium and glutamate-dependent cell death is imminent. Importantly, the ischemic threshold depends on both the regional CBF and duration of cellular hypoxia secondary to this decreased blood flow. For example, a CBF of 18 to 23 mL/100 g/min can be tolerated for 2 weeks, as opposed to 10 to 12 mL/100 g/min for 3 hours and 8 mL/100 g/min for only 1 hour before neuronal death ensues.
In addition to autoregulatory cerebral vascular failure, the injured brain also suffers from uncoupling of cerebral metabolism. In normal brain, cerebral metabolic demand and regional blood flow fluctuate in a proportional manner. Neural activation leads to increased cerebral metabolic activity, which increases the demand for glucose and oxygen and is met by local increases in CBF. This phenomenon is called metabolic autoregulation, with the interaction between metabolic fluctuation and alterations in ICP intimately intertwined at the precapillary level. The close coupling between metabolic supply and demand can be monitored and clinically applied to managing brain-injured patients by correlating cerebral metabolic rate of oxygen consumption (<SPAN role=presentation tabIndex=0 id=MathJax-Element-2-Frame class=MathJax_Error style="POSITION: relative" data-mathml='CMRO2′>[Math Processing Error]CMRO2) and the arteriovenous difference in oxygen saturation (<SPAN role=presentation tabIndex=0 id=MathJax-Element-3-Frame class=MathJax_Error style="POSITION: relative" data-mathml='AVDO2′>[Math Processing Error]AVDO2) as expressed by the Fick equation: <SPAN role=presentation tabIndex=0 id=MathJax-Element-4-Frame class=MathJax_Error style="POSITION: relative" data-mathml='CMRO2′>[Math Processing Error]CMRO2 = CBF × <SPAN role=presentation tabIndex=0 id=MathJax-Element-5-Frame class=MathJax_Error style="POSITION: relative" data-mathml='AVDO2′>[Math Processing Error]AVDO2 or <SPAN role=presentation tabIndex=0 id=MathJax-Element-6-Frame class=MathJax_Error style="POSITION: relative" data-mathml='AVDO2′>[Math Processing Error]AVDO2 = <SPAN role=presentation tabIndex=0 id=MathJax-Element-7-Frame class=MathJax_Error style="POSITION: relative" data-mathml='CMRO2′>[Math Processing Error]CMRO2/CBF. In healthy brain parenchyma, <SPAN role=presentation tabIndex=0 id=MathJax-Element-8-Frame class=MathJax_Error style="POSITION: relative" data-mathml='AVDO2′>[Math Processing Error]AVDO2 is constant, and changes in demand are met by changes in CBF by adjusting local CVR. In the traumatized brain, however, mismatch of supply and demand in the face of preexisting abnormal pressure autoregulation can lead to <SPAN role=presentation tabIndex=0 id=MathJax-Element-9-Frame class=MathJax_Error style="POSITION: relative" data-mathml='AVDO2′>[Math Processing Error]AVDO2 that may be either higher or lower than necessary.
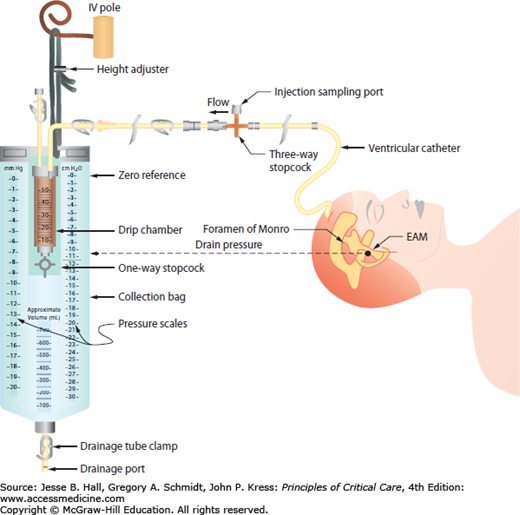
Under physiologic conditions, a MAP of 80 to 100 mm Hg and an ICP of 5 to 10 mm Hg can lead to a CPP of 70 to 85 mm Hg.9 However, the true regional CPP may vary by as much as 27 mm Hg from measurements utilizing global MAP and ICP values.8 Obtaining accurate MAP measurements for CPP determination requires the placement of an arterial pressure catheter, with its transducer at the level of the foramen of Monro, which approximates to the level of external auditory meatus (see Fig. 86-8). The ICP should be measured in units of mm Hg to accurately calculate CPP. In the normal brain, CBF is constant as long as the MAP is maintained between 50 and 150 mm Hg. The local regulation of arterial vascular resistance is affected by CO2, O2, pH/lactate levels, adenosine, nitric oxide, and other components. Neurogenic regulation of the cerebral arterial tone is also regulated by sympathetic input leading to mild tonic vasoconstriction and allowing for higher limits to be reached on the regulation curve.7
FIGURE 86-8
Ventricular drainage system with ICP monitor. Schematic diagram delineating the external ventricular drainage (EVD) system with ventriculostomy ICP monitor. The external auditory canal (approximating the level of the foramen of Monro) is used as a convenient landmark for zeroing the device.
Clinically important factors that influence CBF and ICP are presented in Table 86-2. Control of these factors constitutes the basis for much of the medical management of raised ICP in brain injury. For example, CVR changes linearly within a <SPAN role=presentation tabIndex=0 id=MathJax-Element-10-Frame class=MathJax_Error style="POSITION: relative" data-mathml='PaCO2′>[Math Processing Error]PaCO2 range between 20 and 80 mm Hg. As a result, <SPAN role=presentation tabIndex=0 id=MathJax-Element-11-Frame class=MathJax_Error style="POSITION: relative" data-mathml='PaCO2′>[Math Processing Error]PaCO2 and its manipulation have a dramatic effect on CBF and ICP even when CPP is held constant by alterations in MAP. As an example, inhalation of low CO2 concentrations (5%-7%) can double CBF through changes in extracellular pH that lead to vasodilation of cerebral vasculature and a resultant increase in ICP. To the contrary, low CO2 created by hyperventilation results in vasoconstriction and lowered ICP and can ultimately result in brain ischemia due to prolonged vasoconstriction. Changes in <SPAN role=presentation tabIndex=0 id=MathJax-Element-12-Frame class=MathJax_Error style="POSITION: relative" data-mathml='PaO2′>[Math Processing Error]PaO2 also affect CBF when values fall to less than ∼50 mm Hg, which is demonstrated in Figure 86-9.
FIGURE 86-9
Relationship of cerebral blood flow and 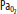 ,
, 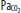 , and CPP. The graph shows the effect of changes in gas tension on cerebral blood flow (CBF), cerebral perfusion pressure (CPP), arterial oxygen partial pressure (
, and CPP. The graph shows the effect of changes in gas tension on cerebral blood flow (CBF), cerebral perfusion pressure (CPP), arterial oxygen partial pressure (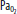 ), arterial carbon dioxide partial pressure (
), arterial carbon dioxide partial pressure (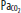 ). CBF is unchanged until arterial oxygen tension (
). CBF is unchanged until arterial oxygen tension (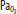 ) falls below about 7 kPa (53 mm Hg) while CBF is proportional to arterial carbon dioxide tension (
) falls below about 7 kPa (53 mm Hg) while CBF is proportional to arterial carbon dioxide tension (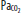 ), subject to a lower limit, below which vasoconstriction results in tissue hypoxia and reflex vasodilation, and an upper limit of maximal vasodilation. CPP is proportional to mean arterial pressure; autoregulation keeps CBF normal as long as CPP is between 50 and 150 mm Hg.
), subject to a lower limit, below which vasoconstriction results in tissue hypoxia and reflex vasodilation, and an upper limit of maximal vasodilation. CPP is proportional to mean arterial pressure; autoregulation keeps CBF normal as long as CPP is between 50 and 150 mm Hg.
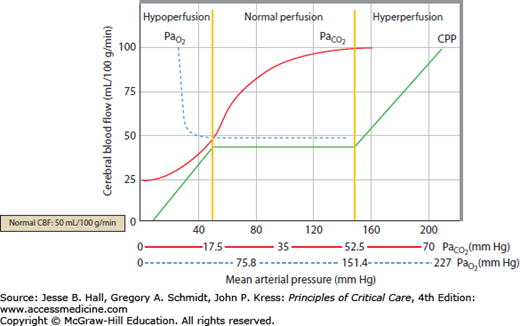
Although the optimal CPP for each individual may vary, it is recommended that in healthy subjects CPP be maintained above 50 mm Hg to avoid ischemia and less than 110 mm Hg to avoid breakthrough hyperperfusion and cerebral edema. Current traumatic brain injury (TBI) guidelines suggest that the optimal CPP in head trauma resides between 50 and 70 mm Hg as CPP levels above 70 mm Hg failed to improve outcome and caused a fivefold increase in acute respiratory distress syndrome.6,10 The presence of a U-shaped curve between CPP and measures of intact cerebral autoregulation, such as PRx and Mx (see below), identifies that both inadequate and excessive CPP are associated with failure of autoregulatory mechanisms. When CPP drops below 40, autoregulation fails and blood vessels collapse (Fig. 86-7), lowering both intracranial blood volume and CBF. Conversely, CPP sustained above 110 mm Hg overcomes mechanisms of autoregulation and hyperperfuses the brain due to passive, irreversible dilation of arterioles with resultant elevation in brain swelling and ICP. Maintenance of CPP within a targeted range of 50 to 70 mm Hg can therefore be an important therapeutic strategy in providing a margin of “reserve” with which the brain can compensate for challenges of normal perfusion via fluctuations in ICP or MAP.
As pressure autoregulation and microcirculatory homeostasis may be severely disrupted in the brain-injured patient, ischemia can result even in the presence of adequate ICP and CPP. Adjusting the target CPP in a particular patient based on the clinical situation, the underlying etiology of brain injury and the vasoreactive state is therefore necessary. Some studies support the concept of CPP targeting based on cerebral vasoreactivity monitoring.11,12 In addition, improved tools for ICP measurements (ie, via minimal invasive intraparenchymal devices) with continuous CPP determinations and the ability to correlate additional brain monitoring parameters such as cerebral blood flow, oxygenation, and chemical profiling allow multimodal, real-time pathophysiologic analysis of brain injury at the bedside.
One of the most feared complications of intracranial hypertension and poor intracranial compliance is the development of plateau waves (PW) (Fig. 86-4). These waves are associated with acute elevations in ICP ranging from 50 to 100 mm Hg. They typically occur in patients with reduced intracranial compliance discussed later. Plateau waves can last from several minutes to more extended durations in severe cases and are rapid in onset and offset. While there are many causes of PW, one important and common mechanism is generalized cerebral vasodilation from an uncontrolled autoregulatory response to a decrease in systemic blood pressure.13 Other causes include processes that increase CBF and CBV (Table 86-2). Since compromised CPP can play an important role in the occurrence of the most severe PW, relative CPP drops should be avoided and/or rapidly treated. Similarly, during a PW, maneuvers that aim to correct CPP toward the target range, such as swift blood pressure augmentation, will potentially abort the PW in many circumstances. Even if blood pressure augmentation does not abort the process, it will likely reduce cerebral ischemia until other treatment modalities can successfully lower the uncontrolled ICP.
CEREBRAL EDEMA, MASS EFFECT, BRAIN HERNIATION
Cerebral edema is an increase in tissue water content within and or around brain cells. Patients with acute brain injuries invariably present with different degrees of edema as a result of different mechanisms of intra-and extraaxial injury. The consequences of uncontrolled edema range from cerebral ischemia to mechanical compression of brain tissue. Initially, edema affects a regional area and can progress to compartmental parenchymal shifts in response to trajectories of increasing pressure differentials. The end result of untreated, progressive brain edema is herniation and ultimately brain death. Despite the obvious clinical importance of cerebral edema, the precise mechanisms of water transport and accumulation of excess water within the brain remain unclear. A series of recent studies on cerebral edema focused on the glial water protein channel aquaporin-4 (AQP4), among others, such as AQP1 and AQP9, that have been shown to facilitate astrocyte swelling (“cytotoxic edema”) and also to be responsible for the reabsorption of extracellular edema fluid (“vasogenic edema”). Therefore, AQP4 modulation via pharmacologic interventions has become an interesting potential therapeutic approach.14-16 AQP4 knock-out, or disruption of its polarized expression pattern, mitigates brain water accumulation and therefore decreases associated ischemia, water intoxication, and hyponatremia in animal models.17
The most common types of cerebral edema are cytotoxic edema from cellular injury and swelling, and vasogenic edema from breakdown of the blood-brain barrier and interstitial fluid extravasation. Other types, such as hydrocephalic edema, ischemic edema (a combination of cytotoxic and vasogenic edema), osmotic edema, and hydrostatic or interstitial edema have also been characterized as distinct entities based on their underlying mechanisms and the predominant location of fluid.18-20 Table 86-3 lists the categories of cerebral edema along with their distinguishing characteristics. Clinically, vasogenic and cytotoxic edema are most frequently encountered. Disruption of the blood-brain barrier results in plasma-derived, protein-rich exudate accumulating in the extracellular white matter, constituting vasogenic edema. Despite the commonly encountered severity of vasogenic edema, CBF is often unaffected and cellular mechanisms remain intact. Among the disease entities with predominant but variable degrees of vasogenic edema are brain tumors (Fig. 86-10), abscesses, traumatic brain injury, and meningitis. Corticosteroids play a primary role in reducing this type of edema, and their effect is most profound when vasogenicity is the primary etiology, as with brain tumors, and to a lesser degree with abscesses.21 In comparison, osmotic agents have little beneficial effect on vasogenic edema.18 Cytotoxic edema, in contrast, is characterized by intracellular swelling of neurons, glia, and endothelial cells with an accompanying reduction in the extracellular space. It occurs without disruption of the blood-brain barrier, and is primarily due to cellular energy depletion, which results in failure of the ATP-dependent sodium pump and accumulation of sodium and water within cells.18 Cytotoxic edema can occur in both gray and white matter. Hypoperfusion (ischemic) injuries are most classically associated with cytotoxic edema. While edema in TBI was thought to be vasogenic in origin, clinical and experimental studies indicate that cytotoxic edema predominates following TBI.22-24 This may explain why drugs that attenuate vasogenic brain edema (eg, corticosteroids) are only beneficial in certain conditions (eg, tumors) but not in others (eg, TBI).25,26 Neuroimaging modalities can aid in characterizing the predominant underlying cause of brain edema such as ischemia, infarction, CSF obstructions, etc.
Classification of Cerebral Edema
| Vasogenic | Cytotoxic | Ischemic | Hydrostatic | Hydrocephalic (Interstitial) | Osmotic | |
|---|---|---|---|---|---|---|
| Pathophysiologic mechanism | Increased vascular permeability | Cellular failure | Anoxia/hypoxia | Increased blood pressure | Impaired CSF outflow or absorption | Relative plasma hypoosmolarity |
| Location | Extracellular | Intracellular | Intracellular and extracellular | Extracellular | Extracellular | Intracellular and extracellular |
| Site | White | White or gray | White and gray | White and gray | White (preferentially periventricular white matter) | White and gray |
| Blood- brain barrier | Disrupted | Intact | Disrupted | Disrupted | Intact | Intact |
| Disorders (examples) | Primary or metastatic brain tumor, Inflammation | Cerebrovascular disorders, fulminant hepatic failure, disequilibrium syndrome | Hypoxic-anoxic encephalopathy | Dysautoregulatory response Posterior reversible edema syndrome (PRES) | Obstructive hydrocephalus | Myelinolysis |
It is important to differentiate mass effect and brain tissue displacement (BTD) from other causes of intracranial hypertension. Displacement of brain tissue may occur in any direction within the cranial vault but is most commonly seen in lateral movement across or along the falx, that is, from one hemisphere across the midline toward the contralateral hemisphere, or as rostrocaudal movement through the tentorial opening and foramen magnum as illustrated in Figure 86-11. Mass effect and resultant tissue shift or herniation can occur without significant elevation in ICP. This depends on the anatomical location of the displaced tissue, whether CSF outlets are obstructed, and whether the volume of the space-occupying lesion causing tissue shift is large enough to overcome compensatory mechanisms. Regional mass effect can cause brain damage through local effects on brain perfusion and exertion of direct mechanical injury to tissue in the absence of globally elevated ICP. Brain tissue displacement can cause depressed consciousness through distortion of key anatomical structures responsible for arousal and attention (diencephalon and brainstem) in the absence of global increases in ICP. Similarly, while reduced consciousness can be the result of elevated ICP secondary to tissue displacement and herniation, it may be seen as a late sign following earlier symptoms relevant to the direct compression of underlying structures. For example, autonomic changes can occur early with evolving BTD, and their recognition can serve as a warning sign of impending herniation.27 The clinical consequences of BTD depend on the etiology, location, size of the lesion, and duration of the process. Table 86-4 summarizes different clinically important herniation syndromes.
FIGURE 86-11
Schematic summary of herniation pathways. Pathways of brain tissue displacement from an expanding supratentorial mass with (1) uncal/lateral, (2) central, (3) subfalcine, (4) transdural/transcranial herniation, as well as (5) ascending transtentorial and (6) tonsillar herniation as seen with an infratentorial mass.
Herniation Syndromes
| Herniation Syndromes | Mechanism/Imaging Findings | Bedside Examination/Comments |
|---|---|---|
| Predominantly Lateral Type | ||
Subfalcine or cingulate herniation |
|
|
Uncal herniation |
|
|
Lateral hemispheric herniation |
|
|
| Transdural/transcranial herniation |
|
|
| Descending Type | ||
Central or bilateral downward transtentorial herniation |
|
|
Tonsillar |
|
|
| Ascending Type | ||
Transtentorial |
|
|
The degree of tissue displacement, and therefore a prediction of compressed structures and expected symptoms, can be approximated by measuring the horizontal shift of the calcified pineal gland on a noncontrast head CT. In a classic study describing this important relationship, horizontal shift of the pineal gland from its midline position (eg, Fig. 86-12B) by 0 to 3 mm correlated with wakefulness; 3 to 5 mm with drowsiness; 6 to 8 mm with stupor; and >8 mm with coma.28 In comparison, rostrocaudal displacement leads to herniation of brain structures through the foramen magnum with resultant pressure on the dorsal brainstem and obstruction of CSF outflow providing two mechanisms for depressed consciousness: direct injury to anatomic structures responsible for arousal and elevated ICP via hydrocephalus. Midline shift is often measured as the distance from the falx (midline) to the septum pellucidum and a ratio can be calculated (Figs. 86-12 and 86-13).29
FIGURE 86-12
Decompressive hemicraniectomy and brain herniation. Axial views of unenhanced head CT imaging after right MCA infarction and decompression (hemicraniectomy) with some transdural brain (release) herniation through the craniectomy defect. Some residual (A) uncal herniation (A) and mild persistent pineal shift (B) and compression of right ventricle (D) are identified. Unfortunately, despite early decompression with small hemicraniectomy site, subfalcine herniation with associated bilateral ACA infarction (C) occurred and some findings of brainstem injury were evident on examination.
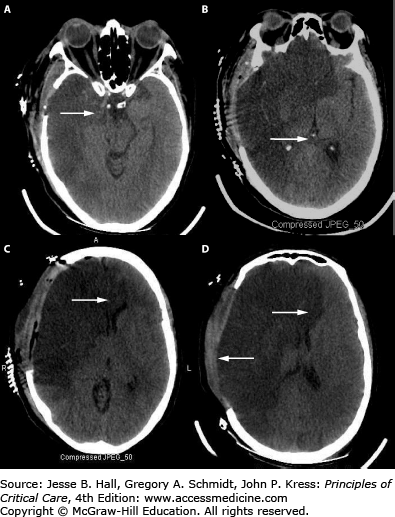
FIGURE 86-13
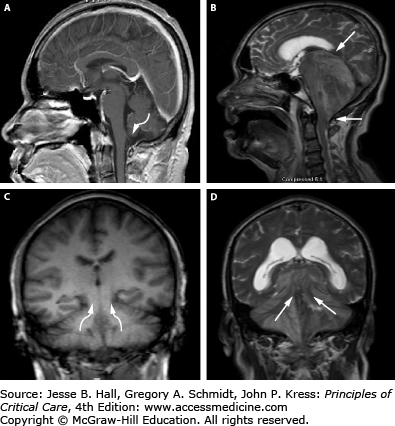
Herniation pathways with infratentorial mass lesions. T1-weighted MRI of a normal brain (A and C) and T2-weighted abnormal posterior fossa lesion (B and D) are presented. Sagittal views (A and B) with arrow indicating the typical pathway for downward cerebellar (tonsillar) herniation into the foramen magnum and ascending transtentorial herniation is shown (B), while the coronal views (C and D) clearly demonstrate the direction of ascending transtentorial herniation through the opening. Acute cerebellar mass lesions (ie, tumors) can readily induce these cerebellar herniation syndromes as well as lead to brainstem compression, obstructive hydrocephalus, and ischemic infarctions from posterior inferior cerebellar artery (PICA) and superior cerebellar artery (SCA) compression at the foramen magnum and tentorial edge, respectively.
Notably, the extent of horizontal shift seen on imaging is not always the primary mechanism of a patient’s reduced level of consciousness. Other differential etiologies should always be considered. Further, the clinical sequelae of tissue displacement and herniation vary greatly among patients due to underlying factors that alter reserve and compliance. As an example, an atrophic brain with significantly increased subarachnoid spaces possesses a greater ability to compensate for tissue displacement by decreasing subarachnoid space to maintain a constant pressure. As a result, an individual’s reserve factors contributing to compliance must be taken into consideration when assessing the relationship between radiographic and clinical presentations in the setting of intracranial mass lesions and tissue displacement. Some sequelae of herniation are summarized in Table 86-4.
Some of the deficits that occur in association with herniation evolve in a predictable manner depending on the location of the primary vector of force of the mass lesion. The falx cerebri (Fig. 86-1) is a dural structure that divides the left and right hemispheres along a sagittal plane. The anterior cerebral arteries course inferiorly and parallel to the falx cerebri and supply blood to the anterior, inferior, and medial frontal lobes. With anterior BTD, brain parenchyma herniates under and across the falx (subfalcine herniation). The anterior cerebral arteries are compressed, placing their territory of supply (mostly the inferomedial frontal lobes and caudate nucleus) in jeopardy (Fig. 86-12). Furthermore, subfalcine herniation (Fig. 86-10C) can lead to obstruction of CSF outflow of the lateral ventricles via compression of the foramen of Monro, resulting in hydrocephalus and elevated ICP. Since subfalcine herniation is a process involving the anterior hemispheres, the patient may not experience depressed consciousness unless shift is extreme or CSF pathways are obstructed.
The tentorium cerebelli is the dural structure that divides the supratentorial compartment, containing the cerebral hemispheres, from the infratentorial compartment, containing the brainstem and cerebellum (Fig. 86-1B). The space between the lateral midbrain and the medial border of the tentorium cerebelli (Fig. 86-11) is called the tentorial incisura, and the posteromedial temporal lobe sits just above this space. A hemispheric mass or brain swelling can force the inferomedial temporal lobe through the tentorial incisura and into the tentorial opening (called lateral transtentorial or uncal herniation) (Fig. 86-11-1). Commonly, this leads to compression of the posterior cerebral arteries (PCA) as they originate from the basilar artery and course around the midbrain (Fig. 86-1). Brain tissue in the PCA distribution, including the occipital lobes, medial temporal lobes, and thalami, are in jeopardy for infarction in this syndrome (Fig. 86-12D). The most common feature of uncal herniation is pupillary enlargement, a sign of third nerve and/or midbrain compression from a medially displaced uncus, which subsequently evolves to stupor, coma, and contralateral pupil dilation from further brainstem compression if the source of the herniation is not corrected. Pupillary changes can reverse with successful, rapid ICP normalization. Uncal herniation can result in trapping of the ipsilateral temporal horn of the lateral ventricle with resultant CSF obstruction, dilation of the temporal horn and surrounding tissue (Fig. 86-12).
Central transtentorial herniation (Fig. 86-11-2) is most common with global, bihemispheric processes (eg, global ischemia/infarction, meningitis, or fulminant hepatic failure) and it classically occurs as the cerebral hemispheres and basal ganglia exert downward pressure, causing brain displacement through the tentorial incisura bilaterally with the pressure cone into the brainstem. If progressive, it results in severe brainstem compression and ischemia with hemorrhage. Bilateral PCA compression can occur with resulting ischemia of the PCA territories as well as the potential for CSF outflow obstruction with hydrocephalus.
In comparison to supratentorial lesions, a posterior fossa mass exerts direct pressure on the brainstem from downward displacement of the cerebellar tonsils (tonsillar herniation) and lower brainstem (medulla) through the foramen magnum (Fig. 86-11-6). This causes severe brainstem and upper cervical spinal cord compression, as well as obstruction of CSF outflow resulting in hydrocephalus. Clinically, these patients develop symptoms of brainstem dysfunction such as autonomic disturbance, altered respiratory patterns, pyramidal tract signs, and cranial nerve palsies as well as depressed consciousness. In addition to downward displacement, posterior fossa lesions can also force cerebellar tissue upward through the tentorial incisura, leading to compression of upper cerebellar and brainstem structures as well as bilateral superior cerebellar artery (SCA) infarctions (Fig. 86-11-5).30 MR imaging (sagittal and coronal views) demonstrates ascending transtentorial and tonsillar herniation secondary to a mass lesion (Fig. 86-13B and D).
As outlined earlier, herniation can occur even without significant measured ICP elevation. Continuous clinical examination and serial brain imaging are therefore needed in addition to ICP monitoring to detect progressive shift. As an example, an acute middle cranial fossa process such as a traumatic temporal hematoma can cause uncal herniation with symptoms of local injury, such as cranial nerve disorder, without a profound rise in ICP. In certain cases, ICP-directed medical treatments can exacerbate BTD; for example, placing an EVD into a trapped ventricle contralateral to a hemispheric mass lesion can increase lateral herniation by relieving opposing CSF pressure. Therefore, we stress the importance of integrating continuous monitoring of several modalities, that is, clinical, ICP, imaging, and other neuromonitoring tools to accurately assess the status of the patient.
When a patient at high risk for brain swelling is encountered, a proactive approach to management should be initiated. This includes a monitoring strategy for early detection of secondary injury caused by edema, mass effect, brain herniation, and any other sources of ischemia. Proactive monitoring of these variables, therefore, provides the best means of detecting and correcting them, preemptively avoiding secondary injury. As mentioned before, monitoring methods include the physical examination, radiographic assessment, and invasive ICP monitoring. ICP monitoring as close as possible to the site of injury should be considered especially when the risk of brain swelling is very high and serial examination or imaging cannot be performed properly (eg, intubated, heavily sedated, difficult to transport).
Serial neurological bedside examination is still the most important, indispensable, and readily available method of examination. The primary drawback to this process, however, is that it can be limiting in detecting changes in brain function and raised ICP. Altered mental status may be due to many etiologies not limited to elevated ICP, and this distinction is impossible to make based on physical examination alone. The patient’s risk factor profile, activity at onset, and tempo of symptom onset and progression limit the differential diagnosis, that is, the rapid onset of acute headache, nausea, vomiting, and altered consciousness in a hypertensive patient indicates intracranial bleeding versus the slow progression of a focal neurological deficit evolving to generalized depressed consciousness in a middle-aged individual pointing toward a primary intracranial tumor. Historical information is essential for an accurate diagnosis and treatment plan; however, in many patients neuroradiological imaging will determine the final diagnosis. In general, headaches from dural stretch of cranial nerve V (trigeminal) sensory fibers, together with intractable nausea and vomiting, followed by neurological deficits including visual disturbances associated with late changes in level of alertness are all typical manifestations of increasing ICP (see Table 86-5 and Fig. 86-14).
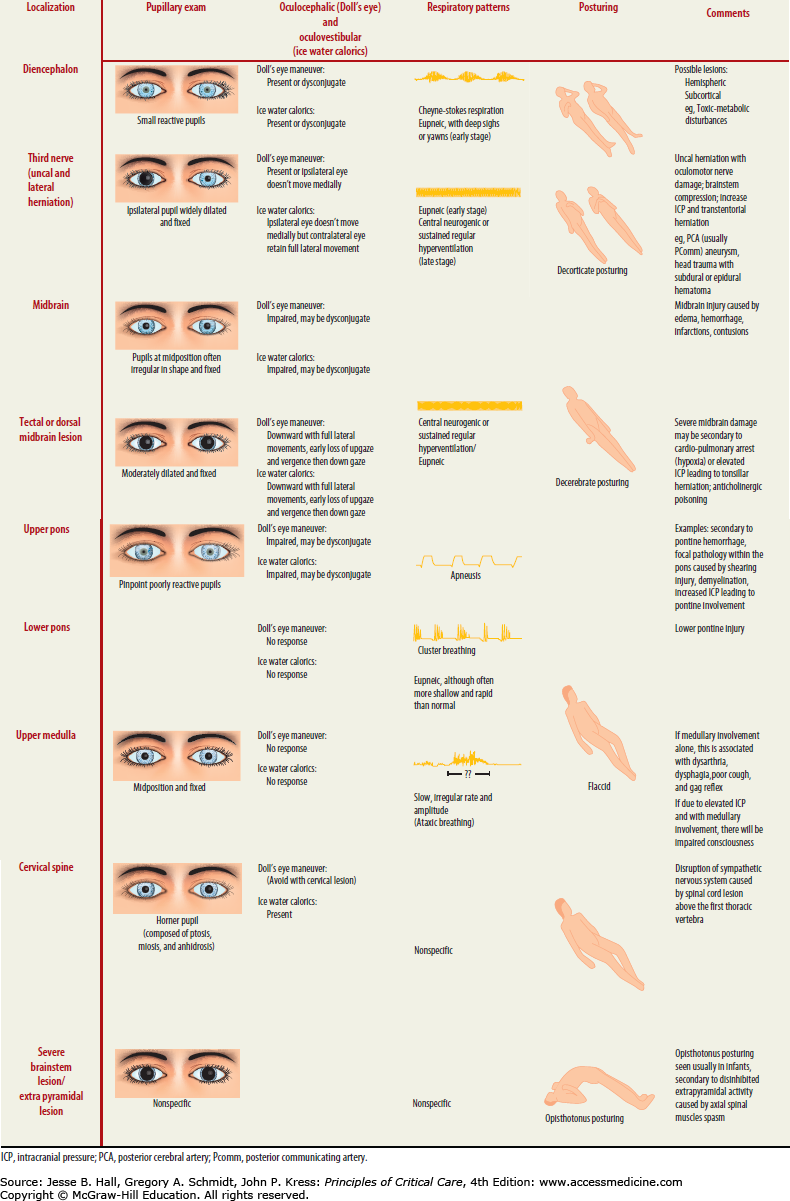
FIGURE 86-14
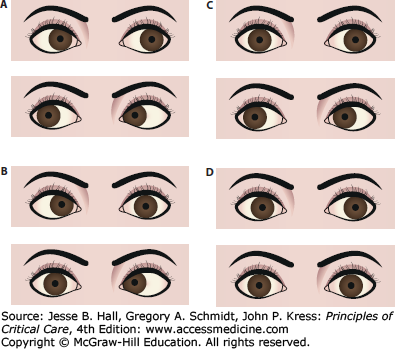
Oculocephalic examination in coma. Once it has been determined in a comatose patient that the cervical spine is stable, the vestibuloocular reflex should be tested by turning the head. With intact brainstem, the eyes will move conjugately away from the direction of turning (that is, the patient has positive doll’s eyes) (A). The doll’s maneuver can also help identify eye abduction deficits as seen in (bilateral) sixth nerve palsies, that is, from hydrocephalus (B), adduction deficits as in brainstem (medial longitudinal fasciculus) lesions leading to internuclear ophthalmoplegia (C), or absent of any lateral (absent doll’s eyes) or horizontal reflex eye movements (D) as seen with severe brainstem injury.
When performing a neurological examination, it is important to use bedside vital sign monitoring as part of the autonomic nervous system evaluation, that is, to identify Cushing triad indicating increased ICP, which consists of systolic hypertension, vagal bradycardia, and respiratory irregularity commonly presenting as irregular tachypnea. Only approximately one-third of patients demonstrate all signs of the triad. Careful observation of the breathing pattern can help define whether ICP is the etiology of the abnormality and can localize the level of injury (Table 86-5). This “autonomic survey” and search for any spontaneous patient movements is often followed by the assessment of a patient’s level of arousal. There are several scales and terms to classify the level of consciousness (Table 86-6). The Glasgow Coma Scale provides a rapid and universal language when describing the degree of brain injury and this classification system should be a component of any intensive care physician’s diagnostic tool set.
Level of Consciousness
| Level | Other Names | Description |
|---|---|---|
| Conscious | “Normal” | Spontaneously awake and alert, promptly stating name, location, date, and time (oriented to three spheres) |
| Confused | Disoriented; impaired thought processing and responsiveness; “clouding of consciousness” | Slow in response with memory time loss, confused, disoriented, difficulty following instruction, delayed responses |
| Delirious | Disoriented; marked loss of attention, restless, illusions, hallucinations, delusions | Also mixture of episodic agitation, somnolence, and obtundation with restlessness or agitation, marked deficits in attention and concentration |
| Somnolent | Drowsiness, state of near-sleep | Dozes after stimuli; incoherent mumbling or disorganized movements observed but still able to follow simple commands upon stimulation |
| Obtunded | Mentally dulled, decreased alertness and psychomotor responses | Decreased interest in surroundings, slowed responses, only brief arousal, unable to follow any commands |
| Stuporous | “Nonspontaneous” | Responds to noxious or painful stimulus by grimacing or pulling away |
| Comatose | “Unresponsive” | Variable examination from intact brainstem reflexes and posturing to complete lack thereof |
Following the assessment of the level of alertness and cognitive function other findings indicative for elevated ICP should be sought (eg, abnormalities of the cranial nerves, motor, and peripheral reflex examinations). Papilledema, defined as edema of the optic nerve that extends anteriorly and laterally into the vitreous humor, is an important and reliable manifestation of raised ICP. It may be asymptomatic in its early stages, but when sustained inevitably progresses to enlargement of the blind spot, blurring of vision, visual obscurations, and ultimately total loss of vision. It usually develops over days to weeks, and is therefore not a manifestation of acute intracranial hypertension in patients with head injury. In a study of patients with head trauma, 54% of patients had increased ICP, but only 3.5% had papilledema on fundoscopic examination.31 Fundoscopic examination reveals loss of venous pulsation, venous engorgement, optic disc hemorrhage, increased diameter of the optic nerve head, and blurring of its margins at the optic disc. Paton lines, defined as circumferential peripapillary retinal folds caused by inward buckling of the swollen disc, are another manifestation of optic nerve edema secondary to raised ICP.
In general, pupillary dilation is seen as a result of third nerve compression identified by pupillary dilation with ptosis, sparing abduction, and intorsion-depression. A third cranial nerve palsy can be seen as a result of a variety of intracranial processes, including uncal herniation in which the nerve is compressed against the tentorium. It can also be seen in the absence of intracranial hypertension as with aneurysms of the posterior communicating artery that directly impinge on the nerve (Fig. 86-1B). In this case, however, the patient does not exhibit additional signs of raised ICP such as altered sensorium, lethargy, nausea, or vomiting. Conditions that exert mass effect on or produce shearing injury to the midbrain structures harboring the third nerve nucleus, such as tumors and trauma, may also produce third nerve palsies without elevated ICP.
Sixth nerve palsies are also seen with elevated ICP as this nerve follows a long intracranial path and is subject to stretching. This palsy is identified by a deficit in ocular abduction. As such, this examination finding is typically a nonlocalizing sign and can only narrow the differential to a process that is elevating ICP. The mechanism, however, cannot be elucidated from this finding. (It can be seen in increased ICP from obstructive hydrocephalus, that is, CSF absorption block after subarachnoid hemorrhage, where there is traction of the subarachnoid segment of the sixth cranial nerve from downward pressure of the pons related to CSF outflow obstruction.) The fourth and seventh cranial nerves are not typically associated with common findings in the setting of elevated ICP.
Other findings in herniation are abnormalities of respiration, which can range from apnea to hyperpnea with undulating crescendo-decrescendo patterns, but also include complete irregularity of breathing with erratic pauses increasing to terminal apnea. As intracranial hypertension escalates, central downward herniation worsens to involve the lower cranial nerves indicating further progression ultimately leading to brain death. This is evident on examination by progressive loss of brainstem reflexes associated with these cranial nerves. At this stage, patients are generally comatose with only minimal brainstem reflexes remaining. Dysfunction of the fifth cranial nerve nucleus within the pons results in loss of the corneal reflex. Disruption of the vestibulocochlear nerve nucleus results in loss of the oculocephalic reflex, which manifests as “doll’s eyes” or the inability to maintain eye position as the head moves. Dysfunction of the ninth and tenth nerves results in loss of lower cranial nerve reflexes including the gag and cough reflex. These are signs of raised ICP in either an acute or more chronic setting, and examination of these reflexes is crucial in assessing the unresponsive patient with suspected elevation in ICP.
Moving to examination of the extremities, specific patterns of posturing and reflexes indicate underlying intracranial hypertension and specific herniation syndromes. Ipsilateral hemiparesis indicates the possibility of uncal herniation causing compression of the contralateral cerebral peduncle as temporal lobe tissue extrudes between the ipsilateral tentorium and the brainstem. This is called “Kernohan notch”. It is therefore a false localizing sign as the hemiparesis is ipsilateral to the site of injury and herniation. For example, a left-sided subdural hematoma causing temporal herniation pushes the opposite cerebral peduncle against the right tentorial edge (right-sided Kernohan notch), leading to a hemiparesis on the same side as the hemispheric lesion.
Posturing, illustrated in Figure 86-15, defined as involuntary flexion or extension of the arms or legs elicited by a painful stimulus, is an important indicator of the amount of brain damage that has occurred secondary to elevated ICP and herniation but also any generalized process resulting in brain injury (eg, shearing or diffuse anoxia). Posturing is therefore included in the Glasgow Coma Scale as a measure of the severity of brain injury and the potential for recovery. There are three types of posturing depending on the level of injury—decorticate (flexor), decerebrate (extensor), and opisthotonos (body arching along the craniospinal axis) posturing (see Table 86-5). These reflexive movements can be spontaneous or stimulus induced. As the level of injury descends to involve progressively increasing portions of the brainstem, posturing movements progress from decorticate to decerebrate. The defining anatomic site that separates these movement patterns is whether the injury is above or below the red nucleus in the midbrain. Flexion or extension can be seen either unilaterally or bilaterally, and can involve one or all extremities. Posturing may even be intermittent.
FIGURE 86-15
Abnormal posturing. Flexion and extension posturing can be seen with or without patient stimulation. Decorticate (top) posturing is due to cerebral lesions with disinhibition at the level of the midbrain (red nucleus) while decerebrate posturing (bottom) is recognized in patients with additional upper brainstem injury secondary to lesions also involving the upper brainstem. At times, a combination of both decorticate and decerebrate posturing can be observed.
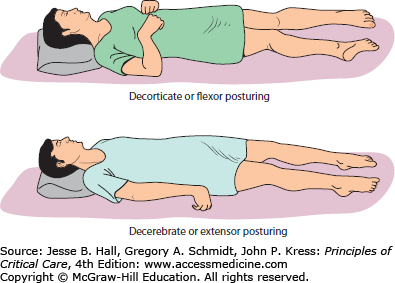
Decorticate posturing (Fig. 86-15) is identified by flexed arms drawn inward to the chest and clenched fists while the legs are extended and rotated inward. It is also referred to as decorticate rigidity, decorticate response, or flexor posturing. Pathophysiologically, flexor posturing represents an acute imbalance between an interrupted corticospinal tract above the mesencephalic red nucleus level, which is unable to relay signals to spinal motor neurons. The subsequently disinhibited red nuclei with increased rubrospinal (and uncontrolled medullary reticulospinal tract) output leads to flexion activity for upper cervical motor neurons. For the lower extremities, the pontine reticulospinal and the medial and lateral vestibulospinal tracts present a disinhibition bias in favor of lower extremity extension over flexion. Therefore, decorticate posturing commonly indicates damage of the cerebral hemispheres, the internal capsule, and the thalamus, possibly also involving the uppermost brainstem.
Decerebrate posturing (Fig. 86-15), also called decerebrate response, decerebrate rigidity, or extensor posturing, is described as involuntary extension of upper and lower extremities, the hallmark being elbow extension. Often, the head is arched back and the patient is rigid with clenched teeth. Decerebrate posturing indicates brainstem damage below the level of the red nucleus due to midbrain distortion secondary to central downward herniation or brainstem compression from cerebellar lesions. Progression from decorticate posturing to decerebrate posturing is often indicative of progressive transtentorial brain herniation due to untreated intracranial hypertension. Opisthotonic posturing (Table 86-5) is an infrequently encountered sign of brainstem injury identified by severe head, neck, and spinal column hyperextension leading to a bridging or arching position of the body that can be seen with severe brainstem injury or extrapyramidal lesions involving the axial muscles.
An important point in the examination of patients with suspected intracranial hypertension is that the degree of abnormal pressure and craniocaudal herniation progresses over time if untreated or if refractory to treatment, leading to alterations in the examination that correlate to higher ICP and more severe neurologic dysfunction. A patient’s examination will therefore change over time, either involving more or fewer of the above findings as ICP is either normalized or worsens. As a result, frequent reexamination is necessary to assess the patient’s response to therapeutic measures. Increasing downward pressure leads to dysfunction first at the level of the diencephalon (ie, thalami), next affecting the upper and middle sections of the brainstem (midbrain and pons), and ultimately impeding medullary function. In summary of the above examination methods, assessment of clinical signs tracks the descending progression of injury in evolving intracranial hypertension. The first is assessment of the respiratory pattern. Respiration patterns become progressively more abnormal depending on the level of injury, evolving from a Cheyne-Stokes pattern to ataxic respirations and then eventually to apnea. The pupils become increasingly more abnormal, initially with some early constriction, then increasing dilation and diminished reactivity, to ultimately becoming fixed, unreactive, and middilated (Fig. 86-14). Reflex eye movements (ie, doll’s eye maneuver) are eventually lost (pontine compromise) (Fig. 86-14 and Table 86-5). Motor responses evolve from localizing to nonlocalizing withdrawal followed by decorticate and then decerebrate posturing and finally flaccidity of all extremities (Table 86-5). The end result of untreated, progressive rostrocaudal brain herniation is brain death with loss of all cranial nerve reflexes and no respiration in the setting of elevated CO2.
Depending on the level of coma or sedation, the patient may not react to any stimulus, or may respond to noxious or painful stimuli by grimacing, grabbing, or withdrawing the stimulated body part. Patients should be stimulated in all extremities to compare the response on both sides in order to attempt to localize the etiology of brain dysfunction. If a patient’s response is equivalent throughout all extremities, it is likely that the etiology is a global insult. If there is a focal response, this finding can help refine the anatomic location of the injury as discussed above. Sensory stimuli may be delivered by pinching a small skin area at the mediolateral forearm or inner thigh; sometimes, however, nail bed pressure is needed. These maneuvers should be avoided in patients with a coagulopathy; as an alternative, pressure applied over the supraorbital notch, bilateral mastoids, or cervicospinal muscles can be used. Most elegantly, some patients respond strongly and reproducibly to intranasal stimulations with a cotton swab.
Frequent bedside examination by physicians and nurses looking for these abnormal findings should be performed in all patients at risk for ICP elevations and the results tracked hourly in the patient’s chart.
INTRACRANIAL PRESSURE MONITORING
ICP monitoring can be an extremely important tool in managing patients in acute brain injury and suspected intracranial hypertension as it provides information on a minute-to-minute basis. The challenges associated with invasive monitoring are the selection of appropriate patients, and the accurate analysis of the information provided by the probe. As with all invasive monitors, the waveform and values provided by an ICP probe should be carefully interpreted, as inaccurate analysis of pressure waveforms can be potentially dangerous. Of note, both the value of the ICP and its waveform provide important information and furthermore, the waveform can indicate worsening pressure dynamics via the Lundberg waves, information that may not be communicated by the ICP value alone.
Invasive ICP monitoring should be used in the setting of a clinical examination that raises concern for intracranial hypertension or a mechanism of brain injury or radiographic findings that would be consistent with elevated ICP, especially in a patient who is unable to be appropriately examined due to sedation or paralysis. In such patients, “blind” management without an ICP monitor can actually exacerbate secondary injury and result in a worse outcome for the patient. For example, uncontrolled use of hyperventilation and osmotic therapy without ICP guidance may lead to decreased CBF and resultant regional or global ischemia.
Regarding appropriate selection of patients for invasive ICP monitoring, the best ICP guidelines are found in the latest Brain Trauma Foundation recommendations (level II evidence). These guidelines indicate the use of ICP monitoring in patients with TBI who remain comatose after resuscitation and if the admission CT reveals intracranial pathology such as hematoma, contusions, or brain edema.32 Generally accepted indications for monitor insertion are processes associated with progressive elevation of ICP such as rapidly expanding intracranial masses secondary to ischemia, hematoma, hemorrhagic tumor, obstructive and nonobstructive hydrocephalus, or diffuse axonal injury (DAI). In all of these forms of brain injury, treatment demands active preservation of stable CPP and ICP to maintain adequate brain perfusion. Neuroimaging studies assist in determining the indication for ICP monitoring. For example, identification of midline shift, effacement of the basal cisterns, or extensive edema helps narrow the differential diagnosis to a process involving elevated ICP. Significant ICP elevations may occur without, or with only subtle, brain imaging findings. Therefore, imaging studies should always be interpreted together with the clinical findings and brain monitoring information. ICP monitoring indications are listed in Table 86-7.
Conditions Often Requiring ICP Monitoring
| Conditions |
|---|
|
To understand the potential benefit of ICP-based treatment algorithms in TBI, Chesnut and colleagues33 prospectively studied 324 patients in Bolivia and Ecuador using random assignment to manage severe TBI patients (GCS 3-8), based on either serial CT imaging and clinical examination (ICE) only or ICE plus invasive ICP monitoring (keeping ICP <20 mm Hg). There was no significant difference in the primary outcome, a composite measure based on percentile performance across 21 measures of functional and cognitive status. Mortality at 6 months was similar—41% versus 39% (p = 0.60), as was median length of ICU stay and distribution of serious adverse events. However, the number of brain-specific ICU treatment days (eg, use of hyperosmolar fluids and hyperventilation) was lower in the ICE plus ICP than the ICE group (3.4 vs 4.8 days; p = 0.002). Taken together, these results seem to support the lack of superiority of an ICP treatment algorithm over treatment solely guided by ICE only in severe TBI patients. The authors concluded that although there were no outcome differences, the qualitative (not quantitative) nature of the ICE-only approach and the increased treatment efficiency (ie, tailoring osmotherapy) in the ICE plus ICP group should not change the practice of ICP monitoring in areas where this resource is available. Further, the authors mentioned that their findings do not argue against the use of ICP monitoring as only the monitoring-based interventional algorithm was tested in their study.
Four important discussion points should be considered prior to reading the study results and the impact on current and modern ICP management strategies:37 the study location and scope of practice in Bolivia and Ecuador; physicians’ expertise and complication rates; variations in ICP interpretation and management skills; variations among the severe TBI patients; and, lastly, the monitoring device employed and the fact that other reasonable indications for ICP monitoring exist. First, it is very likely that the scope of clinical practice depends on location, and more decentralized areas within South America are likely to be dissimilar to those found in North America and Western Europe. De Silva et al showed that 6-month outcome among severe traumatic brain injuries was associated with a higher (51%) mortality in low- and middle-income countries compared to the mortality in high-income countries (30%).36,37 Important treatment differences between continents as well as between individual South American centers and among patients (eg, quality of prehospital stabilization efforts) may have induced significant bias in addition to contributing to worse overall outcomes. Furthermore, initial hospital emergency care and access to rehabilitation were not considered in the study.
Second, even though the intensivists treating the study participants would routinely manage severe TBI patients, there was a lack of prior experience and skills in inserting ICP monitors, dealing with ICP equipment, interpreting and trending the ICP values, understanding ICP waveform morphologies, and correlating ICP findings with imaging and clinical results. Furthermore, the time from primary injury to placement of ICP monitor was not considered, and some patients may therefore have already suffered from secondary brain injury on inclusion in the study. In addition, there is variability in the decision-making process and surgical management of elevated ICP, that is, immediate surgical decompression via hemicraniectomy versus isolated placement of intracranial pressure monitor. Importantly, the trial did not integrate brain tissue oxygen tension, cerebral blood flow monitoring, brain temperature modulation, and other treatment modalities commonly used in modern neurocritical care units to treat severe TBI patients. Also, monitoring for vasospasm in the setting of subarachnoid hemorrhage38 or contusion was not performed. As many as one-third of all severe TBI patients can develop arterial vasospasm detected on TCD or CTA, and its incidence and risk for ischemia can readily abolish an ICP treatment efficacy given the rather small study sample. In addition, the use of a universal, absolute ICP treatment threshold may not be of such great importance as integrating the ICP values to obtain optimal CPP. Of note, CPP-targeted therapy (ie, between 60 and 70 mm Hg) has been shown to be of high importance in improving outcome in moderate to severe TBI as mortality takes on a U-shape form for values below and above this range. The protocol used in the study under discussion was to raise the MAP and/or decrease ICP ≤20 mm Hg but not necessarily targeting specific CPP goals as recommended.
Third, the degree of brain injury among the study patients may already have been severe enough that potential improvements based on ICP monitoring would not impact outcome. Future research could focus on identifying distinct subgroups of severe TBI patients who are likely to benefit from multimodal brain monitoring with optimizing ICP and other brain parameters. Lastly, the device used to measure pressure in the study participants was the intraparenchymal ICP monitor, unlike the external ventricular drains used in many American and European NeuroICU settings. Ventricular drains not only allow CSF drainage to reduce ICP, but also measure ICP in the center of the skull, closer to important brainstem and diencephalic structures, reducing artifact and missed ICP elevations more commonly seen with the more superficial hemispheric measurements of intraparenchymal monitors. In this study, all efforts were directed toward lowering pressure within the cranium, but clinical outcome in survivors also reflects involvement of specific areas of compression, notably, the upper midbrain, thalamus, and reticular activating system.
Other recent systematic reviews on ICP monitoring34 emphasize that the outcome of severe TBI patients depends on guideline-driven management integrating various monitoring elements, and demonstrates that utilizing an ICP monitor alone does not result in better clinical outcome. In comparison, a study by Barmparas et al35 showed that decreased use of ICP monitoring in trauma patients was associated with increased mortality. An explanation for these variable results is that alteration of a single parameter (eg, ICP) may not be expected to significantly impact overall outcome. We recommend that ICP should be treated as an important vital sign but that its values must be carefully integrated into the moment-to-moment clinical and coparameter settings. The concept of managing patients focused on “one ICP value fits all” may only be a part of a more complex strategy in dealing with complicated cases such as acute brain injuries; therefore, management should be tailored to the specific requirements of the individual patient making use of other multimodal monitoring.38
Related posts:






Full access? Get Clinical Tree


