KEY POINTS
Measurement of electrolyte-free water clearance is extremely useful in understanding the pathophysiology of hyponatremia and hypernatremia.
Treatment of hyponatremia should be guided by the degree of symptomatology, rather than the magnitude of the hyponatremia per se.
Hyperkalemia should be treated emergently if typical electrocardiographic (ECG) changes are present; hence, ECG monitoring is indispensable in this setting
Hypocalcemia need only be treated urgently if it is symptomatic.
Severe hyperphosphatemia is seen in the setting of renal failure and/or massive cell lysis.
Total body phosphate stores may be significantly reduced and produce organ dysfunction even in the face of normal or minimally decreased serum levels; if suspicion for a depleted state exists, treatment should be given.
Severe hypomagnesemia may have significant consequences itself, including cardiac arrhythmias and muscle weakness; lesser degrees of hypomagnesemia often accompany hypokalemia and hypocalcemia and correction of the magnesium deficit facilitates correction of the other electrolyte abnormalities.
SODIUM
Sodium is the chief extracellular cation and is critical to regulating extracellular and intravascular volume. Total body sodium determines clinical volume status, but sodium concentration does not correlate with volume status. Both hypernatremia and hyponatremia occur in the presence of hypo-, eu-, and hypervolemia. The sodium concentration itself is the single ion that best represents serum osmolality; essentially all of the clinically relevant symptoms of dysnatremia are secondary to alterations in osmolality. Hypernatremia is largely synonymous with hyperosmolality, while hyponatremia is generally indicative of hypoosmolality.
Osmolality and tonicity are related but separate concepts. Osmolality measures all of the solutes in solution, while tonicity only includes particles that are unable to cross from the intracellular to the extracellular compartment. It is these particles which are osmotically active, and by drawing water across compartments they may alter cell volume. Sodium and potassium are the primary determinants of tonicity.
Balancing water intake and excretion is the principal means by which the body regulates sodium concentration. This balance is maintained by the effects of thirst and antidiuretic hormone (ADH). Following a water load, osmolality falls and osmoreceptors in the hypothalamus suppress thirst and ADH release. The latter signals the kidney to produce dilute urine to clear the water load. In states of water deprivation (or a solute load), osmoreceptors detect the rise in osmolality and increase thirst and ADH.
The kidney regulates osmolality and sodium concentration by diluting or concentrating urine. Producing dilute urine allows the kidney to clear a water load, raising plasma osmolality.
In order to make dilute urine, multiple criteria must be met:
Tubular fluid must be delivered to the diluting segment of the nephron. This can be ensured in any patient with adequate effective arterial blood volume (EABV) and a normal or near normal glomerular filtration rate (GFR).
There must be intact sodium resorption in the diluting segments of the kidney (ie, the thick ascending limb of the loop of Henle [TALH] and the distal convoluted tubule. Loop and thiazide diuretics are the primary causes of inoperative diluting segments.)
The collecting tubule must be impermeable to water. ADH increases water permeability, so the production of dilute urine requires a lack of ADH.
Concentrating urine allows the kidneys to minimize water loss and compensate for an increase in serum osmolality.
In order to produce concentrated urine, the following conditions must be met:
A hypertonic medullary interstitium draws water from the medullary collecting ducts. The TALH creates and maintains the high concentration of the interstitium. Any factor that antagonizes the TALH (eg, loop diuretics, hypercalcemia, or hypokalemia) will disrupt the production of concentrated urine.
The collecting duct must be permeable to water, allowing water to osmotically flow out of the collecting ducts into the medullary interstitium, concentrating the urine. This is affected by ADH. Thus ADH is required for production of concentrated urine.
Antidiuretic Hormone: ADH plays a crucial role in the concentrating and diluting process. ADH, or vasopressin, is a six-peptide amino acid produced in the hypothalamus and stored in the posterior pituitary gland. Release of ADH follows increases in osmolality or dramatic drops in blood pressure or EABV. An increase in serum osmolality of 1% (2 mOsm/kg) will stimulate ADH, while a similar decrease inhibits release. ADH is less sensitive to changes in blood volume; a loss of 7% to 10% of blood volume is required to release ADH.1 When osmolality suppresses and volume depletion stimulates ADH release, volume effects predominate and ADH is released. Osmolality is a more sensitive ADH stimulus, while volume is a more potent ADH stimulus. ADH is also released in response to a collection of nonosmotic, nonvolemic stimuli (Table 99-1).
Free Water Clearance and Electrolyte-Free Water Clearance: As outlined, the renal excretion or retention of water is central to the regulation of osmolality, so specialized concepts have been developed to model renal water handling. Clearance is a generic term used to quantify solute removal (x) by the kidney. Clearance is an artificial construct that represents the volume of blood that is completely cleared of a substance in a set amount of time (Eq. 99-1). The clearance formula can be manipulated to calculate the clearance of free water, called the free water clearance. Conceptually, urine can be divided into two components: an isotonic and a free water component. The isosmotic component contains all of the excreted solute at the same concentration as that found in plasma; since the solute and water loss occur in the same proportion as found in the body, excretion of this isotonic urine does not affect osmolality. The other component is free water; this is solute-free water and excretion of this compartment raises plasma osmolality. For example: A person makes 1200 mL of urine with an osmolality of 142 mOsm/kg. This urine can be divided into 600 mL of isotonic urine (284 mOsm/kg) and 600 mL of free water. In terms of osmolality, only the 600 mL of free water needs to be considered. The loss of this 600 mL will tend to increase serum osmolality. The case is reversed with concentrated urine. A patient produces 1000 mL of urine with an osmolality of 568 mOsm/kg. This urine can be divided into 2000 mL of isotonic urine (284 mOsm/kg) and a negative 1000 mL of free water. In regard to changes in osmolality only the −1000 mL needs to be considered. The −1000 mL represents water that is added to the body and will decrease serum osmolality. Despite the patient excreting 1000 mL of urine, 1000 mL of fluid have been effectively added to the body by the production of concentrated urine. Equation 99-2 is used to calculate the free water clearance.
EQUATION 99-1. Generic clearance formula. Using conventional units, Ux is in milligrams per deciliter of urine, V is in milliliters of urine per minute, and Px is in milligrams per deciliter of blood. After the units cancel, the equation simplifies to milliliters of blood and represents the quantity of blood that is completely cleared of the substance in 1 minute. Ux, urine concentration of x; V, urine volume over a set time period; Px, plasma concentration of x.
EQUATION 99-2. Free water clearance. The derivation of free water clearance begins with the assumption that urine volume is the sum of the solute clearance (Cosm) and free water clearance. From there, algebraic manipulation results in the free water clearance equation.
Free water clearance allows one to model changes in osmolality, but as described above, changes in tonicity and associated alterations in cell volume cause the clinical symptoms of dysnatremia. In order to model changes in tonicity (ie, sodium) rather than osmolality, the free water clearance is further refined to measure electrolyte-free water clearance (CEFW). In electrolyte-free water clearance, serum osmolality is replaced with serum sodium and urine osmolality is replaced with the sum of urine sodium and potassium (Eq. 99-3). To demonstrate the utility of CEFW over <SPAN role=presentation tabIndex=0 id=MathJax-Element-3-Frame class=MathJax_Error style="POSITION: relative" data-mathml='CH2O’>[Math Processing Error]CH2O, consider two patients with identical urine output (800 mL) and urine and plasma osmolality (700 and 270, respectively). One has heart failure and the other has the syndrome of inappropriate secretion of antidiuretic hormone (SIADH). In both patients, the <SPAN role=presentation tabIndex=0 id=MathJax-Element-4-Frame class=MathJax_Error style="POSITION: relative" data-mathml='CH2O’>[Math Processing Error]CH2O is identical at −1274. For every 800 mL of urine they produce, 1274 mL of water is added to the body. When using electrolyte-free water, the two cases look very different. In heart failure (and in all cases of decreased EABV) urine sodium is low, while in SIADH the urine sodium is elevated. Using urine sodium (UNa) = 90, urine potassium (UK) = 60, and plasma sodium (PNa) = 130 for SIADH, the CEFW is −123 mL. Using UNa = 5, UK = 60, and PNa = 130 for congestive heart failure (CHF), the CEFW is 400 mL. In the case of CHF the CEFW is positive, so the kidney is appropriately excreting excess water (although it is limited by oliguria), while in SIADH the CEFW is negative, so the production of urine further lowers plasma osmolality.
EQUATION 99-3. Electrolyte-free water clearance. This formula adapts the free water clearance formula to model changes in tonicity. CEFW, free water clearance; PNa, plasma sodium; UK, urine potassium; UNa, urine sodium; V, urine volume.
Hypernatremia has been reported in 0.2% of hospital admissions and occurs in an additional 0.3% to 1% of patients during their hospital stay.2 These numbers underrepresent the prevalence of hypernatremia in the ICU; a study by Polderman et al showed that 9% of medical ICU patients had a sodium level >150 mmol/L at admission, and an additional 6% developed hypernatremia during the ICU stay.3 More recently, a larger retrospective review of 981 patients showed that 9% of patients admitted to a medical ICU had a sodium level >149 mmol/L, and that hypernatremia developed in a further 2% of patients during their stay.4 The body defends against increases in osmolality and serum sodium by increasing water intake and minimizing renal-free water excretion (through the creation of a concentrated urine). Though ADH is used to minimize free water losses, drinking water alone is able to maintain a normal sodium and serum osmolality despite a total absence of ADH and the production of large amounts of dilute urine. Because the adequate ingestion of water can prevent and reverse hyperosmolality, persistent hypertonicity (and hypernatremia) only occurs when water ingestion is disabled, as occurs with altered mental status, lack of access to water, or inability to drink water. In a review of hypernatremia, 86% of patients lacked access to free water and 94% received less than a liter of electrolyte-free water.2 In the absence of adequate water intake, hypernatremia occurs when CEFW exceeds electrolyte-free water intake. Causes of increased electrolyte-free water clearance are listed in Table 99-2.
Causes of Hypernatremia
| Increased CEFWa | Sweat and Insensible Losses | Water Loss into Cells | Increased Intake of Sodium | Central Impairment of Thirst |
|---|---|---|---|---|
| Central diabetes insipidus | Fever | Severe exercise | Infusions of hypertonic sodium bicarbonate | Reset osmostat |
| Nephrogenic diabetes insipidus | Tachypnea | Seizures | Infusions of hypertonic saline | Elderly patients |
| Hypercalcemia | Burns | Hypertonic dialysate | ||
| Hypokalemia | Exercise | Overdose of salt tablets | ||
| Recovery from acute tubular necrosis | ||||
| Postobstructive diuresis | ||||
| X-linked recessive | ||||
| Lithium | ||||
| Demeclocycline | ||||
| Osmotic diuresis | ||||
| Hyperglycemia | ||||
| Mannitol | ||||
| Urea (catabolic state, high-protein tube feedings) | ||||
| Diarrhea (osmotic) | ||||
| Lactulose | ||||
| Sorbitol | ||||
| Malabsorption |
Etiologies
Loss of Water Electrolyte-free water clearance can exceed water intake either from enhanced renal water loss or extrarenal loss.
Renal Water Loss Renal water losses occur when the kidney is unable to appropriately concentrate urine. This is generically referred to as diabetes insipidus (DI). Diabetes insipidus can be either central or nephrogenic. In central diabetes insipidus (CDI), there is a complete or partial lack of ADH. In nephrogenic diabetes insipidus (NDI), there is a complete or partial end-organ resistance to ADH. Patients with DI have very high CEFW and present with polyuria, polydipsia, and a normal serum sodium. Hyperosmolality and hypernatremia only occur when the patient fails to drink enough water to compensate for the increased CEFW. Renal water loss plays a role in 90% of hospital-acquired hypernatremia, primarily from osmotic diuresis.1
Extrarenal Water Losses The CEFW equation can be modified to look at extrarenal water losses. To do this, change the urinary Na and K to the extrarenal fluid Na and K (see Eq. 99-3). When the fluid Na + K is significantly less than serum Na, electrolyte-free water is being lost, predisposing the patient to hypernatremia. Sweat, osmotic diarrhea, and insensible water loss all result in significant EFW loss.
Use of Hypertonic Fluids The addition of any fluid to the body may alter osmolality. The change in osmolality is predictable using an equation that looks at the volume and electrolyte composition of both the infusate and urine. Equation 99-4 calculates the change in sodium following any combination of infusion and urine production.5 Figures 99-1 and 99-2 show how the change in sodium formula works to predict hypernatremia in two scenarios, one a hypertonic saline infusion and the other an increase in CEFW. The change in sodium formula is a general model of sodium handling in the body and works equally well in both the etiologies and treatment of dysnatremias.
EQUATION 99-4. Change in sodium for any combination of urine and infusate. Viv is the volume of infusate and Naiv is the sodium content of the infusate. Typical values are 0 for 5% dextrose in water, 77 for 0.45% normal saline, 154 for 0.9% normal saline, and 513 for 3% saline; Kiv is the potassium content of the infusate; Vu is the volume of urine; Nau is the sodium content of the urine; Ku is the potassium content of the urine; ∆V is the change in total body volume (Viv − Vu); Nas is the current serum sodium concentration; TBW is the total body water, usually calculated by multiplying weight in kilograms by 0.7 for young men and 0.6 for women and older men.
FIGURE 99-1
Change in sodium following a cardiac arrest. This patient was given 4 amps of sodium bicarbonate during a code. Each amp of bicarbonate contains 100 mL and has a concentration of 1 mmol/mL or 1000 mmol/L. The patient is anuric so the urine volume, Na, and K drop out. The patient weighs 70 kg and has 60% body water so TBW = 42. See caption to Eq. 99-4 for explanation of variables.


Clinical Sequelae: The primary symptoms of hypernatremia are due to loss of cell volume (Fig. 99-3). A decrease in brain volume causes neuromuscular irritability that clinically presents as lethargy, weakness, and headache. These are nonspecific signs and can be particularly occult in the population predisposed to hypernatremia (eg, altered mental status, dementia, and coma). As sodium rises above 158 mmol/L, more severe symptoms may emerge such as seizures and coma, and death may ensue. Interestingly even relatively modest hypernatremia (Na >150 mEq/L) has been associated with increased mortality among ICU patients (RR 1.6) and general inpatients (66% hospital mortality).3,4,6 Significant decreases in brain volume stretch cerebral bridging veins, rendering them susceptible to rupture and intracerebral hemorrhage.7,8 Beyond cerebral effects, hypernatremia inhibits insulin release and causes insulin resistance, predisposing patients to hyperglycemia.
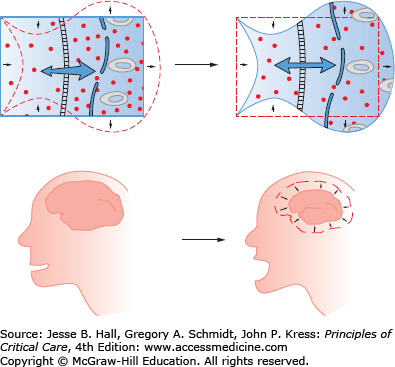
In response to increased serum tonicity and volume loss, cells compensate by increasing the number of intracellular osmoles. Initially cells move extracellular electrolytes into the cells, and later they transfer amino acids and other small molecules into the cell. Increased intracellular osmolality restores intracellular volume, and decreases the clinical impact of hypernatremia.
Treatment: The goal of treating hypernatremia is to arrest any ongoing cause of hypernatremia, and then to restore serum sodium to normal. Specific therapy should be employed to reduce ongoing water losses, such as correcting hypercalcemia-induced diuresis or administering desmopressin (DDAVP) to patients with CDI. Beyond this, correcting hypernatremia requires giving hypotonic fluid either enterally or parenterally. The enteral route is preferred, as it allows the use of electrolyte-free water rather than hypotonic or dextrose-containing fluids. Though the optimum speed of correction has not been rigorously determined, studies on infants and children showed no seizures when sodium was corrected at less than 0.5 mmol/L per hour.9 The sodium can be safely lowered by 10 mmol in the first day of therapy. Patients with acute (<48 h) increases in sodium (eg, from hypertonic bicarbonate infusions) can safely be corrected at 1 mmol/L per hour.10 The change in serum sodium from a given amount of fluid can be calculated using Eq. 99-4. An example of this is shown in Figure 99-4.
FIGURE 99-4
Using the change in sodium formula to assist with the treatment of hypernatremia. In this example the patient is assumed to be anuric. During the treatment urine Na and K should be measured along with urine volume to better refine the estimated volume and time needed to correct the hypernatremia. See caption to Eq. 99-4 for explanation of variables.

A number of complications from the treatment of hypernatremia can occur. If the sodium is lowered too quickly, cerebral edema may occur. Dextrose solutions predispose patients to hyperglycemia, which may cause an osmotic diuresis, worsening the hypernatremia. For this reason, enteral fluids are preferred during treatment of hypernatremia.
Hyponatremia is defined as a serum sodium less than 136 mmol/L. Since serum sodium and its accompanying anions are the principal determinants of serum osmolality, hyponatremic patients are typically hypoosmolar; however, hyponatremia may also be associated with normal or elevated osmolality. Since the primary morbidity from hyponatremia is due to decreased tonicity, hyponatremia with normal or elevated osmolality does not cause the clinical picture typically associated with the more typical hyponatremia with decreased tonicity.
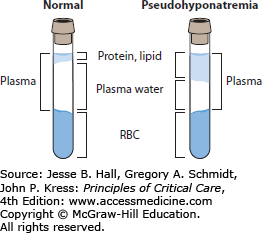
Pseudohyponatremia is associated with a normal plasma osmolality. It is an artifact of two popular sodium assays, flame photometry and indirect potentiometry. Serum with elevated triglycerides, proteins (from multiple myeloma or Waldenström macroglobulinemia, or following intravenous immunoglobulin therapy), or rarely cholesterol has increased solute, and thus a given volume of serum contains less water. This results in a lab error due to overdilution of the sample (Fig. 99-5). Patients suspected of having pseudohyponatremia should have their sodium assessed by direct potentiometry, a technique generally employed by blood gas laboratories that is not susceptible to artifactual hyponatremia. Indirect potentiometry is used in two-thirds of clinical labs, making pseudohyponatremia a real issue.11 Since patients with pseudohyponatremia have normal sodium and osmolality, no specific treatment is needed.
With simultaneous hyponatremia and hyperosmolality, an additional solute is contributing to the osmolality. Both glucose and mannitol can act as the additional solute. The increased extracellular osmolality draws water from the intracellular space, diluting serum sodium. The measured serum sodium falls by a predictable amount; the common cited adjustment is a decrease in sodium of 1.6 mmol/L for every 100 mg/dL increase in blood glucose.12 However, the only empiric data that looked at this showed a more complex relationship: The adjustment of 1.6 mmol/L holds until serum glucose exceeds 400 mg/dL, at which point the sodium should fall by 4.0 mmol/L for every 100 mg/dL rise in glucose. For glucose less than 700 mg/dL, using an adjustment of 2.4 worked nearly as well as the more complex biphasic system.13
Hyposmotic hyponatremia, also called true hyponatremia, and in the remainder of the chapter simply called hyponatremia, occurs when electrolyte-free water intake exceeds electrolyte-free water clearance (CEFW). Intact kidneys are able to clear close to 20 L of electrolyte-free water, so outside of exceptional water intake, hyponatremia only occurs when there is a defect in the CEFW. This defect in CEFW can alternatively be stated as an inability to produce an adequate volume of dilute urine. This can be due to
Decreased delivery of water to the diluting segments of the nephron, namely the thick ascending limb of the loop of Henle (TALH) and distal convoluted tubule (DCT). Decreased delivery of tubular fluid is due to a generalized decrease in glomerular filtration rate (GFR), as seen in renal failure, or increased proximal resorption of water, as seen with decreased EABV.
Decreased activity in the diluting segments of the nephron due to diuretics. Loop diuretics block solute resorption in the TALH and thiazide-type diuretics block resorption in the DCT.
ADH activity, which allows water to be resorbed in the collecting tubules, preventing CEFW.
Etiologies: Hypotonic hyponatremia is traditionally broken down by clinical volume status of the patient (Table 99-3). While this may help clinically classify patients, it does not elucidate the pathophysiology of hyponatremia (eg, CHF and vomiting both cause hyponatremia by inducing a nonosmotic release of ADH, but they are on opposite sides of the classification, as one is hypovolemic and the other hypervolemic). A pathophysiologic approach to hyponatremia categorizes the etiology based on why the patient has compromised CEFW.
Etiologies of Hyponatremia Categorized by Clinical Volume Status
| Hypovolemic | Euvolemic | Hypervolemic |
|---|---|---|
| Diarrhea | SIADH | CHF |
| Vomiting | Hypothyroidism | Cirrhosis |
| Pancreatitis | Glucocorticoid deficiency | Nephrotic syndrome |
| Burns | Acute renal failure | |
| Diuretic induced | Chronic renal failure | |
| Mineralocorticoid deficiency | ||
| Salt-wasting nephropathy | ||
| Cerebral salt wasting |
Decreased Delivery of Water to the Diluting Segments of the Nephron Decreases in GFR for any reason reduce CEFW. Patients with renal failure must moderate their intake of fluids or they may develop acute hyponatremia. Decreases in effective arterial blood volume can result from heart failure, cirrhosis, or volume depletion. Even in situations in which the GFR is intact, decreased EABV (due to CHF, liver failure, or nephrotic syndrome) increases resorption of fluid in the proximal tubule, reducing delivery of fluid to the diluting segments. Patients with reduced distal delivery of fluid have positive CEFW, but the clearance is less than their intake of free water. The hyponatremia tends to be gradual in onset and of mild severity.
Decreased Activity in the Diluting Segments of the Nephron Due to Diuretics An intact diluting segment is essential to CEFW. In some patients, severe hyponatremia can follow the initiation of diuretics.14 Diuretics promote hyponatremia by blocking at least one and possibly all three factors required to produce dilute urine:
Thiazide and loop diuretics both directly antagonize the production of dilute urine.
Diuretic-induced volume depletion reduces the delivery of water to the diluting segments of the nephron.
With more dramatic volume loss, diuretics stimulate a nonosmotic release of ADH, dramatically reducing CEFW.
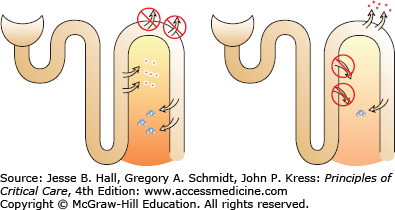
Despite the fact that thiazide-type diuretics are less potent than loop diuretics, they cause hyponatremia more frequently and hyponatremia that is more severe. The NaK2Cl channel, which is antagonized by loop diuretics, is the engine that produces the hypertonic medullary interstitium. This medullary interstitium is necessary for ADH-stimulated water resorption in the medullary collecting duct. Chronic loop diuretic use attenuates the hypertonicity of the interstitium, so that even in the presence of ADH, little water is resorbed (Fig. 99-6).
FIGURE 99-6
In the left panel, thiazide diuretics block reabsorption in the DCT, leaving the TALH intact. ADH-induced water resorption is not dissipated. In the right panel, loop diuretics block TALH so that the medullary interstitium loses its concentration gradient. Even in the presence of ADH, little water is resorbed from the collecting tubule due to the loss of the hypertonic interstitium.
Thiazide-induced hyponatremia is classically seen among elderly Caucasian females.14,15 It should be noted that although these patients are classified as hypovolemic, the volume loss has repeatedly been reported as minor and subtle.16-18 Although hyponatremia associated with diuretic use normally occurs within the first few days of therapy, hyponatremia due to chronic diuretic use may emerge in a newly sick patient.17
ADH Activity The primary role of ADH is to regulate osmolality, so that it is released in response to increases in osmolality. In the presence of hypoosmolality, ADH release is suppressed. However, ADH has a secondary role in maintaining perfusion, and is released in response to large decreases in blood pressure. This nonosmotic release of ADH sacrifices osmoregulation to maintain adequate perfusion. Volume depletion, heart failure, and cirrhosis are all examples in which a nonosmotic release of ADH reduces CEFW and contributes to hyponatremia.
ADH may also be released without osmotic or perfusion-related stimuli. In these cases, the ADH release is considered inappropriate, as it serves no physiologic purpose. The syndrome of inappropriate secretion of ADH (SIADH) is a release of ADH despite decreased osmolality and normal EABV. Causes of SIADH are listed in Table 99-1. Among the elderly no cause of SIADH can be found in up to 10% of patients.19 The diagnosis of SIADH requires four criteria:
Hypotonic (<270 mOsm/kg) hyponatremia (<135 mmol/L)
Inappropriately concentrated urine (>100 mOsm/kg)
Elevated urine sodium
No underlying adrenal, thyroid, pituitary, or renal disease
Unique Clinical Situations Resulting in Hyponatremia
Cerebral Salt Wasting Cerebral salt wasting (CSW) is a rare clinical event that typically follows a subarachnoid hemorrhage. Patients have high urinary flow rates with elevated urine sodium, which ultimately results in severe volume deficiency and hyponatremia. The pathophysiology behind CSW has not been fully elucidated but it is likely that natriuretic proteins are released in response to the CNS injury. Atrial natriuretic peptide, brain natriuretic peptide, and an endogenous ouabain-like peptide have all been proposed as possible etiologic agents. The key diagnostic dilemma is differentiating CSW from SIADH, as both can follow CNS insults and are marked by hyponatremia with elevated urine sodium. The principal differences are urine volume (in SIADH it is low, while it is high in CSW) and clinical volume status (it is normal in SIADH, while it is low in CSW). In CSW, ADH release is secondary to decreased volume status, and aggressive fluid replacement is required to maintain perfusion and suppress ADH. This differentiates it from SIADH, where isotonic saline will decrease serum sodium. CSW spontaneously resolves after 2 to 3 weeks.20
Postoperative Hyponatremia The postoperative period is ripe with factors that stimulate ADH: stress, positive pressure ventilation, pain, nausea, and opioids. It is not surprising that with the addition of generous quantities of IV fluids, hyponatremia can occur.21
A unique form of postoperative hyponatremia may follow urologic or gynecologic procedures employing large amounts of hypotonic irrigants, typically 1.5% glycine.22 The hypotonic fluid enters the systemic circulation and patients develop acute neurologic symptoms from either the rapid drop in sodium or increased ammonia produced from the metabolism of glycine. Patients with hypotonic and neurologic symptoms should be treated for acute hyponatremia. Intact kidneys rapidly clear the irrigant so hyponatremia is only transient. Severe hyponatremia is more common following longer operations, larger resections, and high-pressure irrigation.23
Psychogenic Polydipsia This disorder is most commonly seen in patients suffering from schizophrenia. They overwhelm their urinary diluting capacity by ingesting large volumes of water. Nausea and vomiting from acute hyponatremia are typical and stimulate a nonosmotic release of ADH, worsening the hyponatremia.24 These patients demonstrate excessive diurnal weight gains, usually in excess of 10% of their body weight.
Adrenal Insufficiency Adrenal insufficiency reliably causes hyponatremia. There are two mechanisms that cause this: hypovolemic release of ADH and corelease of ADH with adrenocorticotropic hormone (ACTH). Patients with adrenal insufficiency can be hypotensive due to either loss of cortisol, or in the case of primary adrenal insufficiency loss of aldosterone, causing a salt-wasting nephropathy resulting in hypovolemia. The hypotension and hypovolemia reduces water delivery to the diluting segments and stimulates ADH release. The second mechanism is due to enhanced ADH release in response to increased corticotropin-releasing hormone (CRH). CRH is the secretagogue of ACTH, but it also stimulates ADH release.25,26 In primary and secondary adrenal insufficiency CRH is increased, which will cause increased ADH release, lowering CEFW.
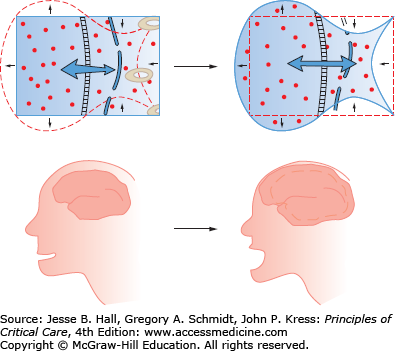
Clinical Sequelae: Symptoms seen in hyponatremia are largely neurologic and increase in severity with lower sodium and increased rate of development of the hypoosmolar state. Symptoms are often vague and nonspecific: malaise, nausea, confusion, and lethargy. More dangerous symptoms follow: headache, obtundation, seizures, coma, and death.16 Unusual symptoms have been reported, including hemiparesis and acute psychosis.27 Symptoms are largely due to cerebral edema (Fig. 99-7). With extracellular hypoosmolality, the intracellular compartment becomes relatively hypertonic and water osmotically flows into cells, resulting in cell swelling and increased intracranial pressure. Following prolonged hyponatremia (24-72 hours), cells compensate for the chronic hyponatremia by ejecting intracellular solutes, lowering intracellular volume. With the restoration of intracellular volume in chronic hyponatremia, the condition becomes essentially asymptomatic.
FIGURE 99-7
Decreases in extracellular osmolality cause the intracellular compartment to be relatively hypertonic. The hypertonic intracellular compartment attracts water, resulting in cellular swelling and tissue dysfunction. In the brain, cellular swelling causes cerebral edema and elevated intracranial pressure.
Hypoxia is repeatedly reported as a common finding among patients with symptomatic hyponatremia. Some authors have attributed this to noncardiogenic pulmonary edema, though central hypoventilation may also be responsible.28 The average partial arterial oxygen pressure (<SPAN role=presentation tabIndex=0 id=MathJax-Element-7-Frame class=MathJax_Error style="POSITION: relative" data-mathml='PaO2′>[Math Processing Error]PaO2) of patients with symptomatic hyponatremia was 63 mm Hg and 68% of the patients in this series were ultimately intubated.29 Hypoxia has been shown to delay cellular compensation, resulting in persistent symptoms of acute hyponatremia despite a prolonged clinical course of over 5 days.
Treatment: Hyponatremia causes symptoms due to cerebral edema from the osmotic movement of water into cells. Compensation for acute hyponatremia consists of cells ejecting intracellular solutes in order to restore normal cell volume. This compensation complicates treatment decisions because rapidly restoring normal osmolality in the presence of compensated cells can cause the serum to be relatively hypertonic to the cells, resulting in the osmotic movement of water out of the cells. In the CNS, this can cause a condition called central pontine myelinolysis (CPM) or osmotic demyelination syndrome (ODS) that results in severe morbidity or death. In determining the treatment plan for hyponatremia, one must balance the risk of cerebral edema from the hyponatremia against the risk of ODS from treating compensated hyponatremia. Creating evidence-based guidelines is difficult because of the lack of randomized controlled trials. Recommendations are based on retrospective case series and expert opinion. The following guidelines attempt to balance the risks of these opposing outcomes.
Symptomatic Hyponatremia In acute hyponatremia, little compensation has occurred and the benefits of rapid correction and resolution of cerebral edema outweigh the risks of ODS. While acute hyponatremia has classically been defined as hyponatremia lasting less than 48 hours, a prospective (albeit not randomized) trial has shown active treatment to be superior to fluid restriction for a cohort of patients with CNS symptoms and an average duration of hyponatremia of 5.2 days (all had hyponatremia for longer than 48 hours and a gradual decrease in sodium of 0.5 mmol/L per hour). Given this, patients with symptomatic hyponatremia regardless of duration should be actively treated. Sodium levels should be initially raised rapidly until symptoms abate or the serum sodium has been raised by 4 to 6 mEq/L.28 Furthermore, the sodium should be raised no more than 10- 12 mEq/L in the first 24 hours, and by no more than 18 mEq/L in 48 hours.30-34 The sodium concentration should be assessed frequently to maintain the permitted rate of correction. Aside from volume depletion, hypertonic saline is the best way to raise the sodium concentration to treat acute, symptomatic hyponatremia. When using hypertonic saline to correct hyponatremia, Eq. 99-4 allows rapid calculation of how much the serum sodium will change in response to 1 L of IV fluid. It should be noted that when using IV fluids to treat hyponatremia, potassium in the IV fluid has the same effect on serum tonicity as sodium, and needs to be added to the sodium.35,36 Figure 99-8 gives an example of using the change in sodium formula to manage hyponatremia.
FIGURE 99-8
Using the change in sodium formula to assist with the treatment of hyponatremia. In this example, the patient is assumed to be anuric. During the treatment urine Na and K should be measured along with urine volume to better refine the estimated volume and time needed to correct the hyponatremia.

Caution should be used when estimating total body water. The common estimate of 0.7 times total body weight for men and 0.6 times total body weight for women assumes a normal hydration status and normal percentage of body fat. This calculation overestimates total body water among patients who are volume depleted or obese. Overestimating TBW leads to exceeding limits on the speed of correction and possibly increased risk of osmotic demyelination.
In the setting of hyponatremia due to volume depletion, normal saline should be used to restore normal perfusion prior to specific therapy for hypoosmolality. Restoring normal perfusion will remove the nonosmotic stimulus for ADH release so the kidney will increase free water clearance and autocorrect the hyponatremia. In some situations, patients will correct their sodium too fast and require free water infusions to slow the rate of correction.
Asymptomatic Hyponatremia Whereas acute symptomatic hyponatremia demands aggressive treatment to reverse cerebral edema, chronic asymptomatic hyponatremia is well tolerated and should be treated conservatively. First, any ongoing cause of the hyponatremia (eg, water intake or diuretics) should be stopped and water restriction initiated. A spot urine sodium and potassium should be checked in order to calculate the CEFW. In most cases, this will be positive and can allow one to determine the degree of fluid restriction required to raise the serum sodium. However, in SIADH the CEFW will be negative, which means that for every milliliter of urine the patient produces, water is added to (rather than cleared from) the body. With a negative CEFW water restriction will rarely be successful at raising serum sodium. In this unique situation, a loop diuretic can increase the CEFW by reducing the urine sodium. A negative CEFW only occurs when the urine Na plus urine K is higher than the serum Na, and loop diuretics typically reduce the urine sodium to around 70, which is sufficient to reduce the urine Na plus K to below serum Na. This will make the CEFW positive and allow fluid restriction to increase the serum sodium. (Note: In patients with a positive CEFW, adding a loop diuretic can have the opposite effect by increasing a low urine Na, indicative of good CEFW, to around 70.)
In patients with chronic SIADH, use of the antibiotic demeclocycline acts as an ADH antagonist and so allows more liberal fluid intake. Likewise, increasing the solute load by using a high-protein diet or increased sodium and potassium intake will also allow increased daily fluid intake.
The Role of Vasopressin Antagonists There are three AVP receptor subtypes (V1a, V1b, and V2). Vaptans are nonpeptide competitive inhibitors of V2 primarily, the receptor subtype which mediates the effects of ADH.37 They reduce urine osmolality, increase CEFW (aquaresis), and consequently increase serum sodium concentration.
Currently there are no data to support the use of vaptans in acute symptomatic hyponatremia.38 However, their use has been examined in chronic hyponatremia from various causes. In the SALT-1 and SALT-2 trials, Tolvaptan was efficacious at raising serum sodium at day 4 and day 30 in patients with euvolemic and hypervolemic hypernatremia.39 Initial studies supported the efficacy of long-term vaptan use in chronic hyponatremia.40,41 However, due to concerns of liver injury the FDA recently advised that use of Tolvaptan should not exceed 30 days, and that its use be avoided in those with underlying liver disease.42 Additional limitations include expense compared to more standard therapies, and the potential for over rapid correction, which is more likely to occur with use of these agents.38 Furthermore, increased thirst may limit the rise in serum sodium.39
Osmotic Demyelination Syndrome ODS is the primary complication of therapy for hyponatremia. With rapid or complete restoration of extracellular osmolality, a well-adjusted intracellular environment becomes relatively hypotonic. Water then flows from the intracellular to the extracellular compartment, causing cell volume collapse. In the CNS, this can cause a demyelinating lesion. Symptoms usually present within a week of the correction of the hyponatremia. Although slowly evolving neurologic symptoms similar to a pseudobulbar palsy and the “locked-in” quadriparesis are classic, findings may be more subtle. Disturbances of movement or behavior or seizures may be the presenting finding.43 Some patients tend to be more susceptible than others. In a case-control study by Ayus and colleagues, patients with hepatic encephalopathy, hypoxia, or normalization of Na (or an increase greater than 25 mmol/L) in the first 48 hours were found to be susceptible to ODS.44 In the event that ODS occurs, there is some evidence that reintroducing hyponatremia improves outcome. In a rat model of ODS, hypotonic fluid administration improved both survival and neurological outcomes. The greatest benefit was observed from early relowering of the sodium.45 In humans with ODS, case reports also suggest benefit of early relowering of sodium.46-48 Some authors advocate relowering the sodium to 120 mEq/L by giving hypotonic fluids and DDAVP, and then allowing the sodium to slowly return to normal. However, there are no randomized trials in the literature to validate this approach, and given the devastating morbidity and mortality of ODS, an attempt to reduce the sodium level to a value of 15-17 mEq/L greater than the initial (lowest) value should be attempted in patients displaying symptoms of ODS.
POTASSIUM
Potassium is the most common cation in the body. The ratio of the intracellular to extracellular potassium concentration is the primary determinant of the resting membrane potential (Em). Alterations in the Em disrupt the normal function of neural, cardiac, and muscular tissues. Normal serum potassium ranges from 3.5 to 5.2 mmol/L. The molecular weight of potassium is 39.1, so a daily potassium intake of 80 mmol is roughly equivalent to 3.1 g of potassium.
The normal physiologic handling of potassium can be viewed as a three-step process: ingestion, cellular distribution, and excretion. Irregularities at any of these steps can result in pathologic serum potassium concentrations.
Intestinal Absorption: Normal daily intake is roughly 40 to 80 mmol. Potassium is rapidly and completely absorbed by the small intestine. Net GI absorption (intake minus GI losses) is approximately 90%.49 Lower GI secretions have high concentrations of potassium, 80 to 90 mmol/L, but due to the limited amount of stool (80-120 g/d), daily GI excretion of potassium is only 10 mEq.50-52 The colonic epithelium is capable of actively excreting potassium, but this is not clinically significant. Patients with chronic renal failure have elevated stool potassium but total potassium excretion is still limited to about 12 mEq/d.53
Cell Uptake: Following absorption, potassium distributes among the intracellular and extracellular compartments. The intracellular compartment acts as the primary buffer to changes in serum potassium concentration.
The Na-K-ATPase pump, driven by a ubiquitous cell surface enzyme, moves potassium into cells while pumping sodium out of cells. The pump is stimulated by β2-adrenergic activity, while α-adrenergic activity results in potassium efflux.53 Insulin also stimulates the activity of this pump and is independent of its hypoglycemic activity.54
Extracellular pH can affect the cellular distribution of potassium. Various explanations have been proposed, including a direct effect of pH on the Na-K-ATPase, or an H+-K+ exchange to maintain electroneutrality. The effect of pH on potassium distribution varies depending on the nature of the acid-base disturbance. Respiratory acidosis, alkalosis, and organic acidosis all have minimal effect on potassium distribution. Inorganic acidosis can increase serum potassium, while metabolic alkalosis can lower potassium.
Renal Excretion of Potassium: Renal excretion of potassium can range from 5 to 500 mEq/d.55,56 Though 500 mmol of potassium is filtered by the glomerulus each day, >90% is resorbed in the proximal tubule and loop of Henle. Thus the secretory contribution from the distal tubule is the main determinant of urinary potassium excretion.57 Because of this phenomenon, the study of renal potassium handling can focus exclusively on the distal nephron.
The secretion of potassium in the distal tubule is governed by two phenomena: tubular flow and aldosterone activity (Fig. 99-9). Potassium secretion by the principal cells of the collecting duct depends on a favorable electrochemical gradient. Rapid tubular flow provides a continuous supply of potassium-depleted fluid, maintaining a favorable chemical gradient. Increased tubular flow occurs with high tubular sodium delivery. Furthermore, resorption of sodium by the principal cells generates a negative charge in the tubular lumen. The luminal electronegativity enhances potassium secretion. Some of this negative charge is lost by concurrent resorption of chloride. Decreased distal chloride delivery, as occurs with metabolic alkalosis, reduces chloride resorption, thereby increasing the tubule electronegativity and enhancing potassium secretion.58 Aldosterone is a steroid hormone produced in the zona glomerulosa of the adrenal gland. Its principal site of action is the connecting segment and collecting tubules of the distal nephron. In the principal cells of the cortical collecting duct (CCD), aldosterone increases the resorption of sodium and hence the secretion of potassium. Aldosterone stimulates the production and activity of Na-K-ATPase, sodium channels, and potassium channels.59 Aldosterone also has a small but measurable effect on increasing GI potassium excretion.49 Aldosterone is secreted in response to angiotensin II and elevated serum potassium.
FIGURE 99-9
Potassium handling in the cortical collecting duct. The resorption of sodium creates a negatively charged tubule lumen. This charge helps drive the secretion of potassium. Chloride resorption decreases the negative charge, so increased chloride resorption decreases potassium secretion. ADP, adenosine diphosphate; ATP, adenosine triphosphate; Pi, inorganic phosphate.
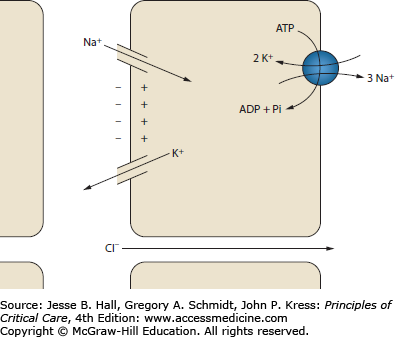
The fact that potassium secretion is dependent on both tubular flow and aldosterone means that urinary potassium excretion is independent of volume status despite the fact that both tubular flow and aldosterone are intimately tied to volume status. With volume depletion, increased angiotensin II stimulates the release of aldosterone, which enhances potassium secretion; however, the simultaneous decrease in GFR and increased resorption by the proximal tubule decrease tubular flow, antagonizing potassium secretion. In the opposite case of volume overload, decreased aldosterone suppresses potassium secretion, but increased tubular flow enhances potassium secretion, maintaining potassium balance.
Hypokalemia is defined as a serum potassium concentration below 3.5 mmol/L, and is found among 20% of the hospitalized population. However, this high frequency probably does not reflect total body potassium depletion. In a review of 70 hospitalized patients with a potassium less than 2.8 mmol/L, the potassium rose toward normal regardless if they were given potassium or not. The authors suggested that hospitalization for acute illness was associated with increased adrenergic stimulation, resulting in intracellular movement of potassium and transient hypokalemia.60
Etiology: See Table 99-4.
Causes of Hypokalemia
| Decreased Potassium Intake | Cellular Shift | Increased Potassium Loss |
|---|---|---|
| Anorexia | β-Adrenergic activity | Extrarenal losses |
| Malnutrition/malabsorption | Endogenous | Chronic diarrhea |
| Alcoholism | Albuterol | Fistulas and ostomies |
| Ingestion of grey clay | Dobutamine | Renal losses |
| Terbutaline | Loop diuretics | |
| Fenoterol | Thiazide diuretics | |
| Insulin | Osmotic diuretics | |
| Alkalemia | Acetazolamide | |
| Periodic paralysis | Type I and II renal tubular acidosis | |
| Thyrotoxicosis | ||
| Familial | Metabolic alkalosis | |
| Xanthines | Bicarbonaturia (vomiting) | |
| Theophylline toxicity | Ketonuria | |
| Caffeine | Hypomagnesemia | |
| Barium toxicity | Carbenicillin | |
| Treatment of anemia (rapid cell proliferation) | Bartter and Gitelman syndromes | |
| Hyperaldosteronism | ||
| Exogenous steroids | ||
| Adrenal adenoma | ||
| Adrenal hyperplasia (Conn syndrome) | ||
| Syndrome of apparent mineralocorticoid excess | ||
| Liddle syndrome | ||
| Congenital adrenal hyperplasia | ||
| Renal artery stenosis | ||
| Renin-secreting tumor |
Decreased Dietary Intake Potassium-poor diets usually are merely contributory to hypokalemia. In a study of normal individuals, a potassium restricted diet (20 mmL/day) was associated with a decline in serum potassium from 4.1 mEq/L to 3.5 mEq/L.61 Even among patients with severe malnutrition due to anorexia nervosa and/or bulimia, serum potassium less than 3 mmol/L occurred in less than 2%, and in all of those patients there was enhanced potassium loss from cathartics or vomiting.62
Cellular Shifts Activation of β-adrenergic receptors increases Na-K-ATPase activity. Any physiologic stress that releases epinephrine or norepinephrine can result in a transient decrease in serum potassium. Use of primarily β-adrenergic catecholamines, such as dobutamine, can cause transient hypokalemia.63 The β-agonists used for bronchodilation or as tocolytic agents can also acutely lower potassium.
Insulin reliably stimulates Na-K-ATPase and lowers serum potassium.54 Insulin-induced hypokalemia has been documented in the treatment of diabetic ketoacidosis and hyperosmolar nonketotic states, and with the use of intravenous dextrose solutions.64 Refeeding syndrome occurs among patients given a carbohydrate-rich diet or parenteral nutrition following periods of starvation. Refeeding syndrome is associated with hypokalemia and hypophosphatemia.65
Metabolic (or respiratory) alkalosis is associated with the intracellular movement of potassium. Increased pH results in movement of hydrogen ions from the intracellular to the extracellular compartment. Potassium shifts into cells to maintain electroneutrality. In addition, in metabolic alkalosis, increased serum bicarbonate enhances renal potassium excretion. Studies in nephrectomized dogs show a modest but measurable decrease in serum potassium of less than 0.3 mmol/L for each increase in pH of 0.1 (though these data did not account for a significant increase in serum osmolality).66 The common association of alkalosis and hypokalemia is primarily due to enhanced renal excretion of potassium rather than a transmembrane shift.
Hypokalemic periodic paralysis is an unusual clinical entity in which transcellular shifts in potassium result in paralysis. These patients develop sudden, severe drops in serum potassium associated with skeletal muscle paralysis. Triggers include carbohydrate loads, exercise, and changes in body temperature. Acetazolamide may decrease the frequency and severity of symptoms in some families; recent work suggests this response varies according to genotype.67,68 Oral potassium can be used to treat acute paralysis but patients often develop rebound hyperkalemia.69
Increased Potassium Loss The cortical collecting duct is the critical site of renal potassium handling. Normally aldosterone activity and sodium delivery to the CCD are balanced so that when one is elevated the other is decreased. Excess renal potassium excretion only occurs when both aldosterone and distal sodium delivery are increased.
Most diuretics increase distal delivery of sodium and increase aldosterone, resulting in hypokalemia. Primary hyperaldosteronism causes hypertension and hypokalemia. The hypokalemia is due to the simultaneous increase in aldosterone activity and sodium delivery to the distal nephron. The increased sodium delivery is due to a spontaneous diuresis in response to the hypertension known as aldosterone escape. A full discussion of the causes of increased aldosterone activity is beyond the scope of this text; however, a list of causes is included in Table 99-4.
Normally, the primary anion in the tubular fluid is chloride. Various conditions can result in chloride being replaced by an unresorbable anion. Anions that are not resorbed prevent sodium from being resorbed and increase sodium and tubular fluid delivery to the distal nephron. In addition, unresorbable anions increase tubule electronegativity, which enhances potassium secretion by the principal cells. The most common example of an unresorbable anion resulting in hypokalemia is bicarbonate. In metabolic alkalosis, increased serum bicarbonate is delivered to the distal nephron, resulting in increased renal potassium loss. Diabetic ketoacidosis increases delivery of the unresorbable anion β-hydroxybutyrate to the distal nephron.
Hypomagnesemia is associated with hypokalemia that is resistant to therapy. Decreased magnesium increases renal potassium losses and needs to be corrected prior to successful treatment of hypokalemia.70
Despite a high concentration of potassium in lower GI secretions, 85 to 95 mmol/L, GI potassium losses are typically modest, about 10 mEq/d.51 Chronic diarrhea can cause hypokalemia, but the mechanism appears to be more complex than simple GI loss of potassium. In cases of experimental diarrhea, daily GI potassium loss was never higher than 24 mEq/d, a level well below average daily potassium intake.71 In addition, studies on diarrhea show that as stool volume increases, stool potassium concentration falls, ultimately reaching a level similar to that of plasma in cases of severe cholera.49 Explanations for the commonly seen association of diarrhea and hypokalemia include secondary hyperaldosteronism, diminished intake of potassium, or transcellular shifts of extracellular potassium.
Gastric secretions have potassium content similar to that of plasma, 5 to 8 mmol/L. Gastric losses result in severe metabolic alkalosis and secondary hyperaldosteronism, both of which enhance renal potassium loss.
Clinical Sequelae: Hypokalemia is a well-known risk factor for a variety of cardiac arrhythmias. Increased ectopy with hypokalemia has been documented in ambulatory hypertensive patients, in patients undergoing coronary artery bypass grafting, and during acute myocardial infarction (AMI).72,73 Following AMI, hypokalemia increases the risk for a number of arrhythmias; patients with hypokalemia are more than twice as likely to develop ventricular fibrillation.74 Hypokalemia enhances the risk of digitalis toxicity and associated arrhythmias. Digitalis-induced arrhythmias may occur despite normal digitalis levels in the presence of modest hypokalemia.75
A drop in extracellular potassium hyperpolarizes the muscle cells, which can prevent myocyte depolarization. Clinically, this can lead to weakness, fatigue, cramping, and myalgia. Severe cases can result in paralysis. Numerous case reports of respiratory muscle weakness and respiratory failure have been reported with hypokalemia due to diabetic ketoacidosis. Severe hypokalemia can cause rhabdomyolysis. Alcoholics may be particularly prone to proximal muscle weakness due to rhabdomyolysis.76,77
Hypokalemia can cause polyuria due to increased thirst and by inducing a mild and reversible renal concentrating defect.78,79 The etiology of the concentrating defect is multifactorial, but primarily represents decreased renal response to ADH.
Gastrointestinal complications are primarily related to decreased gut motility associated with hypokalemia. Serum potassium of less than 3.0 mmol/L is associated with constipation. Paralytic ileus can occur as potassium falls below 2.5 mmol/L.
Hypokalemia stimulates the proximal tubule to increase ammoniagenesis. Patients predisposed to hepatic encephalopathy can develop encephalopathy from this increased ammonia load.80
Diagnosis: Hypokalemia is defined as a serum potassium concentration less than 3.5 mmol/L. Once hypokalemia has been established, the primary diagnostic goal is differentiating renal from extrarenal potassium loss. Urine studies are used to separate extrarenal losses, in which the kidneys are potassium avid, from renal losses, in which the kidney inappropriately wastes potassium. Three studies may be used to differentiate these states: spot urine potassium concentration, 24-hour urine potassium, and the transtubular potassium gradient (TTKG).
The spot urine is the simplest test to use. The urine potassium should be less than 20 mmol/L in the face of hypokalemia. If the spot potassium is greater than 40, renal potassium wasting should be suspected. Urine potassium of 20 to 40 mmol/L is considered nondiagnostic.81 There are two primary problems with this test; the first is it fails to control for changes in the water content of urine. Since hypokalemia is associated with decreased ADH sensitivity, increased water content will lower the urinary potassium concentration. The second problem is that spot samples provide information for only a single moment in time. Patients with diuretic-induced hypokalemia become potassium avid after the diuretic has cleared. One study on the diagnosis of hypokalemia (mean K = 2.0 mmol/L) found spot urine potassium to have a sensitivity of 40% and specificity of 100% for excess renal potassium loss.82
The 24-hour urine potassium test avoids both of the above problems at the expense of increased complexity and a 24-hour delay. Patients with hypokalemia should reduce urinary potassium losses to less than 15 mEq/d. Potassium losses greater than that indicate inappropriate renal losses. The 24-hour urine provides no information on the renal potassium handling prior to the urine collection (eg, diuretic use that is stopped prior to collection will show an appropriately potassium-avid kidney).
The transtubular potassium gradient calculates the ratio of tubular potassium to venous potassium at the end of the CCD. The CCD is responsible for potassium excretion, so increases in the TTKG indicate renal wasting of potassium, while decreases indicate renal potassium conservation (Fig. 99-10 and Eq. 99-5). When serum and renal potassium handling are normal, the TTKG runs from 5 to 8.81,83 In the face of hypokalemia, the CCD should minimize the potassium excretion, resulting in a reduced TTKG. The TTKG has been validated in patients with decreased dietary potassium, periodic paralysis, diuretic-induced hypokalemia, primary hyperaldosteronism, and vomiting.83,84
EQUATION 99-5. The transtubular potassium gradient (TTKG). Plasma Osm, plasma osmolality; urine Osm, urine osmolality.
FIGURE 99-10
The transtubular potassium gradient measures the ratio of tubular potassium to interstitial potassium and quantifies the renal excretion of potassium. ADH, antidiuretic hormone; CCD, cortical collecting duct; DCT, distal convoluted tubule; TALH, the thick ascending limb of the loop of Henle.
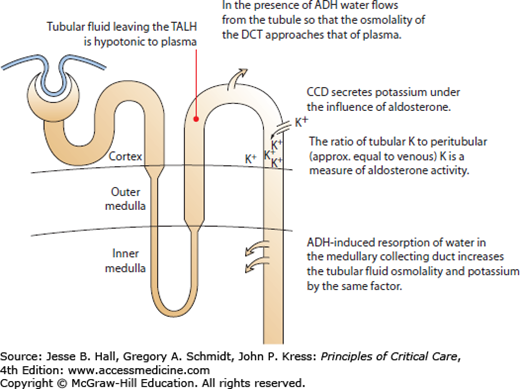
The TTKG has two assumptions that must be met prior to using this formula85:
There must be ADH activity to ensure that the osmolality of the tubular fluid approximates the osmolality of blood by the end of the cortical collecting duct. ADH activity is ensured by only using the formula when urine osmolality exceeds serum osmolality.
There must be adequate tubular sodium to allow the cortical collecting duct to secrete potassium. The test should only be done if the urine sodium concentration is greater than 25 mmol/L.
Treatment: The treatment of hypokalemia can be broken down into three questions: when to treat, with which potassium salt, and with what quantity. The National Council on Potassium in Clinical Practice has published clinical practice guidelines on potassium replacement. The guidelines recommend correcting potassium in any patient with potassium below 3.0 mmol/L and select patients with serum potassium below 3.5 mmol/L. They specified a more aggressive treatment regimen for patients with hypertension, congestive heart failure, and increased risk for or history of cardiac arrhythmias or stroke.86
Determining the dose of potassium to correct hypokalemia is difficult because there is not a firm relationship between serum potassium and total body potassium. Balance studies have shown that potassium is disproportionately lost from the extracellular compartment rather than total body potassium (eg, a 25% drop in serum potassium is due to less than a 25% drop in total body potassium). Sterns and colleagues analyzed the results of seven balance studies and found a linear relationship for potassium deficit and serum potassium (r = 0.893). The loss of 100 mmol of potassium lowered the serum potassium by 0.27 mmol/L, so a fall from 4 to 3 mmol/L represented a 370-mmol potassium deficit.87 In Scribner and Burnell’s review, they estimated that a drop in potassium from 4 to 3 mmol/L represented a loss of 100 to 200 mmol of potassium, and a drop in serum potassium from 3 to 2 mmol/L represented an additional 200 to 400 mmol deficit.88 These estimates do not account for altered cellular distribution of potassium. In diabetic ketoacidosis, for example, serum potassium overestimates total body potassium, while β-agonist–induced hypokalemia underestimates total body potassium. In most cases of hypokalemia due to cellular redistribution, experts advise against treatment, as the hypokalemia is transient and treatment predisposes the patient to hyperkalemia. One exception to this is symptomatic periodic paralysis in which respiratory arrest due to hypokalemia may occur, so emergent treatment is indicated. Caution should still be used, as rebound hyperkalemia is common.
The form of potassium used in repletion is most often potassium chloride. The chloride anion has some advantages over alternatives such as phosphate, bicarbonate, or citrate. The chloride anion is primarily an extracellular anion, which minimizes the movement of potassium into the cell, maximizing the change in serum potassium. Chloride also does not increase the secretion of potassium at the collecting duct. The use of alternate potassium salts should be reserved for specific clinical scenarios in which there is an indication for the anion (eg, citrate in metabolic acidosis and phosphate in hypophosphatemia).
In patients who are asymptomatic, oral replacement is sufficient and doses from 40 to 100 mEq of KCl per day are typically sufficient to correct the hypokalemia over several days.86 Increasing intake of potassium-rich foods is less effective than potassium chloride supplements because the anions associated with dietary potassium are primarily phosphate and citrate.
Potassium chloride can be given as a liquid, in crystalline form (often marketed as a salt substitute), or pills with multiple formulations and coatings. The bioavailability of all these formulations is identical, with greater than 70% absorption.89 The liquid formulation has the fastest absorption and lowest patient compliance of all formulations (due to the bad taste).90 Wax-matrix extended-release tablets are associated with gastrointestinal tract ulcers and stenotic lesions. The microencapsulated extended-release formulations have the best compliance and low rates of GI side effects.90-92
Parenteral potassium should be used to correct symptomatic hypokalemia or when patients are unable to take oral medications. Twenty to forty millimoles of KCl in 0.5-1 L of isotonic saline or 5% dextrose is a typical solution. Saline solutions are preferred as dextrose solutions stimulate insulin release that can result in acute worsening of the hypokalemia.77,93 The use of saline with dilute concentrations of potassium means that patients must get multiple liters of saline to correct even modest potassium deficits, which may be contraindicated in volume-overloaded patients.
Concentrated potassium solutions delivered at a rate of 10-40 mEq/h in smaller volumes are frequently used in the ICU setting. The use of these solutions had generated fears about the possibility of precipitating arrhythmias from local, transient hyperkalemia near the infusion site or by causing peripheral vein irritation from caustic potassium solutions. Despite these concerns, the use of concentrated potassium infusions, 200 mmol/L, at a rate of 20 mEq/h in the ICU was shown to be safe and efficacious in both a retrospective study of 495 infusions and a prospective study of 40 patients.94,95 Twenty milliequivalents of KCl increased the serum potassium by 0.25 mmol/L 1 hour after the infusion finished. The peak rise in serum potassium, 0.48 mmol/L, was at the end of the infusion. ECG monitoring showed no change except for decreased ventricular ectopy. Potassium was infused safely through both peripheral and central sites. Infusion rates of greater than 20 mEq/h are best administered through a central vein.
Hypomagnesemia is a common cause of treatment failure. Patients who are resistant to potassium supplementation should have serum magnesium measured, and if low, repleted. Patients with diuretic-induced hypokalemia often benefit from the initiation of a potassium-sparing diuretic. Amiloride has been shown to mitigate magnesium losses associated with loop and thiazide diuretics.
Patients on amphotericin B often become hypokalemic. Both spironolactone (100 mg twice a day) and amiloride (5 mg twice a day) have been shown to increase serum potassium and decrease the use of potassium supplements in randomized prospective trials.96,97
In patients with recalcitrant vomiting (ie, bulimia) and associated hypokalemia, one treatment strategy is to decrease the loss of hydrogen ions with a proton-pump inhibitor or H2 blocker.98 Proton pump inhibitors may have a similar role in ameliorating hypokalemia associated with gastric suction.
Etiologies: The ability of the kidney to excrete potassium is flexible and adaptable. If dietary ingestion of potassium is increased over a number of days, the kidney increases daily potassium excretion to match. Because of this, dietary loads of potassium do not result in hyperkalemia unless they are sudden, or paired with a defect in renal potassium handling. Likewise, conditions associated with the movement of intracellular potassium to the extracellular space are associated with only transient hyperkalemia because either the kidneys excrete or the cells reuptake the excess potassium. Defective renal potassium handling increases susceptibility to hyperkalemia from increased potassium intake or transcellular shifts (Table 99-5).
Causes of Hyperkalemia
| Increased Potassium Intake | Cellular Shift | Decreased Potassium Excretion |
|---|---|---|
| Oral | β-Blockers | Decreased tubular flow |
| Dietary | Lack of insulin | Renal insufficiency |
| K supplements | Acidemia (inorganic) | Prerenal azotemia |
| Salt substitutes | Digitalis toxicity | Volume depletion |
| Ingestion of red clay | Succinylcholine | Congestive heart failure |
| Enteral feeding supplements | Hyperkalemic periodic paralysis | Cirrhosis |
| NSAID use | ||
| Parenteral | Hypertonicity | Decreased stimulation of aldosterone |
| Medical error | Hyperglycemia | Type IV RTA (hyporeninism) |
| TPN | Mannitol | ACE inhibitor use |
| CVVH replacement | Cell destruction | Angiotensin-receptor blocker |
| fluid | Ischemia | Decreased synthesis of aldosterone |
| Peritoneal dialysis fluid | Necrosis | Adrenal insufficiency, primary |
| Hemolysis | Ketoconazole | |
| Old blood transfusions | Rhabdomyolysis | Heparin |
| Treatment of hypokalemia | Tumor lysis syndrome | Congenital adrenal hyperplasia |
| Penicillin | Chemotherapy | Decreased aldosterone activity |
| (K formulations) | Radiation therapy | Spironolactone |
| Spontaneous | Trimethoprim | |
| Amiloride | ||
| Triamterene | ||
| Cyclosporine A | ||
| Tacrolimus | ||
| Type I RTA, hyperkalemic variety | ||
| SLE, obstruction, sickle cell | ||
| Decreased GI excretion | ||
| Constipation in ESRD patients |
Increased Potassium Intake Dietary potassium is typically in the range of 40 to 80 mEq/d. Hyperkalemia has been reported to follow the use of potassium chloride salt substitutes, even in the presence of normal renal function.99 One teaspoon of potassium chloride contains 50 to 65 mEq of potassium. Enteral nutrition supplements may be rich sources of potassium. Ensure Plus at 100 mL/h provides 130 mEq of potassium per day.
Red blood cell transfusions can have extracellular potassium concentrations as high as 70 mmol/L.100 The risk of hyperkalemia from transfusions rises as the age of the transfusions increases (Table 99-6). Use of “washed” packed red blood cells reduces the risk of transfusion-associated hyperkalemia.101
Potassium Concentration in Red Blood Cell Transfusions
| Age (Days) | Plasma Potassium (mmol/L)a | Extracellular Potassium (mmol) per 250 mL of PRBC (Hematocrit 60%) |
|---|---|---|
| 0 | 1.6 | 0.2 |
| 7 | 17 | 1.7 |
| 14 | 27 | 2.7 |
| 35 | 44 | 4.4 |
| 42 | 46 | 4.6 |
Intracellular Redistribution of Potassium Increases in plasma osmolality, most often due to hyperglycemia, causes an osmotic movement of water from the intracellular compartment. Potassium moves out of the cell with the water. Using mannitol to increase serum osmolality from 283 to 300 mmol/kg increased potassium from 4.4 to 5.2 mmol/L.102
The Na-K-ATPase is critical in preventing intracellular potassium from causing hyperkalemia. Any factor that decreases the activity of this enzyme will cause potassium to leak from cells. A lack of insulin slows the Na-K-ATPase. In diabetic ketoacidosis hyperkalemia is typical despite total body potassium depletion, and in this setting is largely related to the hyperglycemia.
β-Blockers inhibit the Na-K-ATPase activity and are associated with a mild increase in serum potassium. Uremia reduces Na-K-ATPase activity so that renal failure patients are less able to use the intracellular compartment to buffer potassium loads. Digitalis is an Na-K-ATPase antagonist. Digitalis toxicity can cause severe hyperkalemia. Removing digitalis with binding antibodies allows rapid correction of the hyperkalemia.103
Inorganic acids increase serum potassium. Attempts to predict the change in potassium from changes of pH have shown tremendous variability (0.3-1.1 mmol/L for a decrease in pH of 0.1) and are considered unreliable.102 Decreases in pH due to respiratory or organic acidosis have minimal effect on serum potassium.
Cell death results in release of intracellular potassium. Large-scale cell death can cause fatal hyperkalemia. Tissue necrosis and hyperkalemia can be seen with rhabdomyolysis of any etiology. Likewise, tissue ischemia can cause cell death and release large amounts of potassium. Bowel and limb ischemia are occult causes of hyperkalemia. Hemolysis causes hyperkalemia by releasing the intracellular potassium of red blood cells. Tumor destruction with chemotherapy results in release of intracellular contents. Tumor lysis syndrome (TLS) is hyperphosphatemia, hyperuricemia, hyperkalemia, and hypocalcemia associated with acute renal failure (due to uric acid nephropathy). The use of hydration and hypouricemic agents in prophylaxis regimens has substantially reduced the incidence of TLS.104 The syndrome most often occurs with poorly differentiated neoplasms with large tumour burden and/or high proliferation rates such as Burkitt lymphoma and acute leukemias, but it has been reported with breast cancer, medulloblastoma, and ovarian and lung cancer. In some rapidly growing tumors, spontaneous lysis occurs prior to therapy. Hyperkalemia in tumor lysis syndrome is more common in patients with premorbid renal insufficiency.105
Related posts:






Full access? Get Clinical Tree


