KEY POINTS
Acute adrenal insufficiency in critically ill patients is best referred to as critical illness–related corticosteroid insufficiency (CIRCI).
CIRCI may arise due to adrenal insufficiency or tissue resistance to cortisol.
Adrenal insufficiency is best diagnosed by a random cortisol <10µg/dL (Type 1 CIRCI) or a delta of <9µg/dL after a 250µg cosyntropin stimulation test (Type II CIRCI).
The diagnosis of adrenal insufficiency/CIRCI should not be made on the basis of laboratory criteria alone.
Treatment with low-dose hydrocortisone (200 mg/day) or methylprednisolone (60 mg/day) should be considered in patients with septic shock who have responded “poorly” to resuscitation with fluids and vasopressor agents and those with ARDS who have failed to show an improvement within 48 hours of supportive care. The role of low-dose hydrocortisone in patients with severe sepsis and other clinical situations in the ICU remains to be determined.
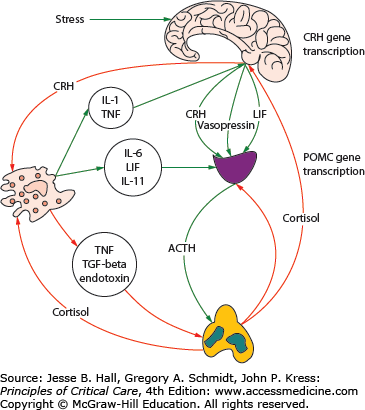
Exposure of the host to diverse noxious stimuli results in a stereotypic and coordinated response, referred to by Hans Selye as the general adaption syndrome (or stress response) which serves to restore homeostasis and enhance survival.1 The stress response is mediated primarily by the hypothalamic-pituitary-adrenal (HPA) axis as well as the sympathoadrenal system (SAS). Activation of the HPA axis results in increased secretion from the paraventricular nucleus of the hypothalamus of corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP). CRH plays a pivotal integrative role in the response to stress. CRH stimulates the production of ACTH by the anterior pituitary, causing the zona fasciculata of the adrenal cortex to produce more glucocorticoids (cortisol in humans).2 AVP is a weak ACTH secretagogue and vasoactive peptide that acts synergistically with CRH to increase secretion of ACTH. The increase in cortisol production results in multiple effects (metabolic, cardiovascular, and immune) aimed at restoring homeostasis during stress. In addition, the HPA axis and immune system are closely integrated in multiple positive and negative feedback loops (see Fig. 102-1). Activation of the SAS results in the secretion of epinephrine and norepinephrine from the adrenal medulla and to an increased production of inflammatory cytokines such as interleukin-6 (IL-6).
FIGURE 102-1
Activation of the hypothalamic-pituitary adrenal axis (HPA) and the interaction with the inflammatory response. ACTH, adrenocorticotrophic hormone; CRH, corticotropin releasing hormone; IL-6, interleukin-6; IL-11, interleukin-11; LIF, leukemia inhibitory factor; POMC, proopiomelanocortin; TGF-β, transforming growth factor-beta; TNF, tumor necrosis factor. (Reproduced with permission from Marik PE, Pastores SM, Annane D, et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med. June 2008;36(6):1937-1949.)
CORTISOL PHYSIOLOGY
Cortisol (hydrocortisone) is the major endogenous glucocorticoid secreted by the adrenal cortex. Over 90% of circulating cortisol is bound to corticosteroid-binding globulin (CBG) with less than 10% in the free, biologically active form. CBG is the predominant binding protein with albumin binding a lesser amount. During acute illness, particularly sepsis, CBG levels fall by as much as 50%, resulting in a significant increase in the percentage of free cortisol. The circulating half-life of cortisol varies from 70 to 120 minutes, with a biological half-life of about 6 to 8 hours. The adrenal gland does not store cortisol; increased secretion arises due to increased synthesis under the control of ACTH. Cholesterol is the principal precursor for steroid biosynthesis in steroidogenic tissue. In a series of sequential enzymatic steps, cholesterol is converted to pregnenolone and then to the end products of adrenal biosynthesis, namely, aldosterone, dehydroepiandrostenedione, and cortisol. At rest and during stress about 80% of circulating cortisol is derived from plasma cholesterol, the remaining 20% being synthesized in situ from acetate and other precursors. High-density lipoprotein (HDL) is the preferred cholesterol source of steroidogenic substrate in the adrenal gland.3
The activities of glucocorticoids are mediated by both the glucocorticoid (GR) and mineralocorticoid receptors (MR). The GR and MR share both functional and structural homology.4 Both aldosterone and glucocorticoid hormones bind to both the GR and MR. At low basal levels cortisol binds to the high-affinity, low-capacity MR. However, with increased cortisol secretion the MR are saturated and cortisol then binds to the low affinity, high-capacity GR. In addition, the 11β-hydroxysteroid dehydrogenase (11β-HSD) enzymes play an important role in preventing glucocorticoid access to cells that express the MR.5,6 This enzyme has two isoforms: an NAD+-dependent form (11β-HSD-2) and an NADP+-dependent form (11β-HSD-1). 11β-HSD-2 is found in tissues with high levels of MR activity such as the kidney, sweat and salivary glands, placenta, and colon. 11β-HSD-2 converts cortisol to cortisone, its inactive reduced metabolite which is unable to bind to the GR and MR. 11β-HSD-1, which is found in glucocorticoid target tissues, catalyzes the conversion of cortisone to the active glucocorticoid cortisol. Proinflammatory cytokines modulate the activity of the 11β-HSD enzymes, with interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) increasing the activity of 11β-HSD-1 while decreasing that of 11β-HSD-2.7,8
Cortisol diffuses rapidly across cell membranes binding to the GR. Two isoforms of the GR have been isolated, namely GR-α and GR-β. The GR-β isoform fails to bind cortisol and activate gene expression and thus functions as a negative inhibitor of GR-α.9 The GR-β binds to the glucocorticoid antagonist RU-486 and may play a role in regulating gene expression.10 Seven isoforms of GR-α have been reported; these isoforms may be selectively expressed by different tissues with each isoform eliciting a distinct response.11,12 Through the association and disassociation of chaperone molecules, the glucocorticoid-GR-α complex moves into the nucleus where it binds as a homodimer to DNA sequences called glucocorticoid-responsive elements (GREs); these are located in the promoter regions of target genes which then activate or repress transcription of the associated genes (see Fig. 102-2). In addition, the cortisol-GR complex may affect cellular function indirectly by binding to and modulating the transcriptional activity of other nuclear transcription factors such as nuclear factor κB (NF-κB) and activator protein-1 (AP-1). Overall, glucocorticoids effect the transcription of thousands of genes in every cell of the body. It has been estimated that glucocorticoids affect 20% of the genome of mononuclear blood cells.13
FIGURE 102-2
An overview of the mechanisms of action of glucocorticoids. CBP, cyclic AMP response element binding (CREB) binding protein; cPLA2α, cytosolic phospholipase A2 alfa; eNOS, endothelial nitric oxide synthetase; FKBP51/52, FK-binding protein 51/52; FYN, FYN oncogene–related kinase; GILZ, glucocorticoid-induced leucine zipper protein; GRα, glucocorticoid receptor α; Grβ, glucocorticoid receptor β; HSP90, heat shock protein 90; LCK, lymphocyte-specific protein tyrosine kinase; MAPK, mitogen-activated protein kinases; MKP-1, MAPK phosphatase 1; NF-κB, nuclear factor kappa B; PI3K, phosphatidylinositol 3-kinase; PKB, protein kinase B; PKC, protein kinase C; POMC, proopiomelanocortin; SLPI, secretory leukoprotease inhibitor. (Reproduced with permission from Marik PE. Critical illness related corticoseroid insufficiency. Chest. January 2009;135(1):181-193.)
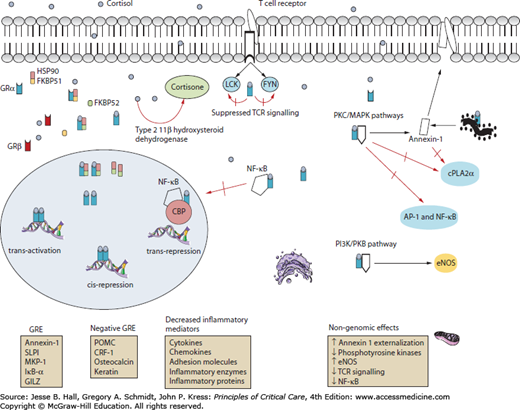
Glucocorticoids play a major role in regulating the activity of NF-κB which plays a crucial and generalized role in inducing cytokine gene transcription.14-16 NF-κB is normally maintained in an inactive form by sequestration in the cytoplasm through interaction with inhibitory proteins (IκBs). Upon stimulation by lipopolysaccharide, double-stranded DNA, physical and chemical stresses, and inflammatory cytokines, the latent NF-κB/IκB complex is activated by phosphorylation and proteolytic degradation of IκB, with exposure of the NF-κB nuclear localization sequence. The liberated NF-κB then translocates to the nucleus and binds to promoter regions of target genes to initiate the transcription of multiple cytokines including TNF- α, IL-1, and IL-6 and cell adhesion molecules (eg, ICAM-1, E-selectin) and other mediators of inflammation. Glucocorticoids inhibit the activity of NF-κB by increasing the transcription of IκBs and by directly binding to and inhibiting NF-κB.15,16
Cortisol has several important physiologic actions on metabolism, cardiovascular function, and the immune system.17,18 The metabolic effects of cortisol include an increase in blood glucose concentrations through the activation of key enzymes involved in hepatic gluconeogenesis and inhibition of glucose uptake in peripheral tissues such as the skeletal muscles. In addition, in adipose tissue, lipolysis is activated resulting in the release of free fatty acids into the circulation. Cortisol also has a permissive effect on other hormones increasing glucose levels, including catecholamines and glucagon. Sustained cortisol hypersecretion stimulates glucose production at the expense of protein and lipid catabolism and insulin resistance.
Cortisol increases blood pressure through several mechanisms involving the kidney and vasculature. In vascular smooth muscle, cortisol increases sensitivity to vasopressor agents such as catecholamines and angiotensin II.19,20 These effects are mediated partly by the increased transcription and expression of the receptors for these hormones.19,20 While the effect of glucocorticoids on nitric oxide (NO) is complex, it appears to increase endothelial nitric oxide synthetase (eNOS), thereby maintaining microvascular perfusion.21-24 Cortisol has potent anti-inflammatory actions including the reduction in the number and function of various immune cells, such as T and B lymphocytes, monocytes, neutrophils, and eosinophils at sites of inflammation. Cortisol decreases the production of cytokines, chemokines, and eicosanoids and enhances the production of macrophage migration inhibitory factor.25,26
CRITICAL ILLNESS–RELATED CORTICOSTEROID INSUFFICIENCY
There has recently been a great deal of interest regarding the assessment of “adrenal function” and the indications for corticosteroid therapy in critically ill patients. While the use of high-dose corticosteroid (10,000 to 40,000 mg of hydrocortisone equivalent over 24 hours) in patients with severe sepsis and ARDS failed to improve outcome and was associated with increased complications,27,28 an extended course of “stress-dose” corticosteroids (200-350 mg hydrocortisone equivalent/day for up to 21 days) has been demonstrated to increase ventilator- and hospital-free days and improve short-term survival in select groups of ICU patients.29-32 These patients typically have an exaggerated proinflammatory response and are considered to be “relatively” corticosteroid insufficient.
Until recently the exaggerated proinflammatory response that characterizes patients with systemic inflammation has focused on suppression of the HPA axis and “adrenal failure.” However, experimental and clinical data suggest that corticosteroid tissue resistance may also play an important role. This complex syndrome is referred to as critical illness–related corticosteroid insufficiency (CIRCI).2,33 CIRCI is defined as inadequate corticosteroid activity for the severity of the patient’s illness. CIRCI manifests with insufficient corticosteroid mediated downregulation of inflammatory transcription factors.
Tissue corticosteroid resistance is a well-known manifestation of chronic inflammatory diseases such as chronic obstructive pulmonary disease (COPD), severe asthma, systematic lupus erythematosus (SLE), ulcerative colitis, and rheumatoid arthritis. Emerging data suggest that corticosteroid tissue resistance may develop in patients with acute inflammatory diseases such as sepsis and acute lung injury (ALI).2,34 In a sheep model of ALI induced by Escherichia coli endotoxin, Liu et al demonstrated decreased nuclear GRα-binding capacity and increased expression of phospholipase A2 (PLA2) despite increased serum cortisol levels.35 These authors demonstrated similar findings in the liver cytosol following a burn injury in rats, which were partially reversed by TNF-α and IL-1β neutralizing antibodies.36 In an ex vivo model, Meduri and colleagues compared the cytoplasmic to nuclear density of the GR complex in patients with ARDS who were survivors and nonsurvivors.34 These authors demonstrated a markedly reduced nuclear density of the GR complex in nonsurvivors while the cytoplasmic density was similar between survivors and nonsurvivors. This experiment provides further evidence that the nuclear GC-GR activity may be impaired in critically ill patients despite adequate cytoplasmic (serum) levels of cortisol.
HPA axis failure appears to be a common problem in patients with systemic inflammation. Patients at risk for developing tissue glucocorticoid resistance are similarly at risk for HPA axis failure. The incidence of HPA axis failure varies widely depending on the criteria used to make the diagnosis and the patient population studied. The overall incidence of “adrenal insufficiency” in critically ill patients approximates 20%, with an incidence as high as 60% in patients with severe sepsis and septic shock.37 The mechanisms leading to inadequate cortisol production during critical illness are complex and poorly understood, and likely include decreased production of CRH, ACTH, and cortisol.2 A subset of patients may suffer structural damage to the adrenal gland from either hemorrhage or infarction and this may result in long-term adrenal dysfunction. In addition, a number of drugs are associated with adrenal failure. However, reversible HPA dysfunction is increasingly being recognized in critically ill patients with systemic inflammation associated with the sepsis, ALI, liver disease, and following cardiopulmonary bypass (see Table 102-1).
Causes of Adrenal Insufficiency
|
CLINICAL MANIFESTATIONS OF CIRCI
Patients with chronic adrenal insufficiency (Addison disease) usually present with a history of weakness, weight loss, anorexia, and lethargy with some patients complaining of nausea, vomiting, abdominal pain, and diarrhea. Clinical signs include orthostatic hypotension and hyperpigmentation (primary adrenal insufficiency). Laboratory testing may demonstrate hyponatremia, hyperkalemia, hypoglycemia, and a normocytic anemia. This presentation contrasts with the features of CIRCI. The clinical manifestations CIRCI are consequent upon an exaggerated proinflammatory immune response. Hypotension refractory to fluids and requiring vasopressors is a common manifestation of CIRCI.17,38 CIRCI should therefore be considered in all ICU patients requiring vasopressor support as well as those with severe progressive ARDS.2 Patients usually have a hyperdynamic circulation which may compound the hyperdynamic profile of the patient with sepsis/systemic inflammation.38 However, the systemic vascular resistance, cardiac output, and pulmonary capillary wedge pressure can be low, normal, or high.39 The variability in hemodynamics reflects the combination of CIRCI and the underlying disease. CIRCI should also be considered in patients with progressive ALI. Laboratory assessment may demonstrate eosinophilia and hypoglycemia. Hyponatremia and hyperkalemia are uncommon.
DIAGNOSIS OF ADRENAL INSUFFICIENCY AND CIRCI
The diagnosis of adrenal insufficiency in the critically ill is fraught with difficulties. Furthermore we have no test that quantifies corticosteroid activity at the tissue level. Traditionally the diagnosis of adrenal insufficiency in the critically ill has been based on the measurement of a random total serum cortisol (“stress” cortisol level) or the change in the serum cortisol in response to 250µg of synthetic ACTH (cosyntropin) the so-called delta cortisol.18,40 Both of these tests have significant limitations in the critically ill.41 Commercially available cortisol assays measure the total hormone concentration rather than the biologically active free cortisol concentration42 Furthermore, the timing of cortisol measurements may be important as large hourly variations in cortisol have been reported.41,43 In addition, the reproducibility of the ACTH stimulation test is poor in critically ill patients.43,44 To complicate the issue further, the specificity, sensitivity, and performance of the commercially available assays are not uniform.45 Despite these limitations, Annane and colleagues have reported that a delta cortisol of less than 9µg/dL was the best predictor of adrenal insufficiency (as determined by metyrapone testing) in patients with severe sepsis/septic shock.37 A cortisol of less than 10µg/dL was also highly predictive of adrenal insufficiency (PPV of 0.93); however, the sensitivity of the test was poor (0.19).
One approach to resolving the question of whether too little glucocorticoid signal ultimately “gets through” is to examine target tissues whose function is regulated in part by glucocorticoids. Through their inhibitory effects on nuclear factor-κβ signaling pathways, glucocorticoids are the most potent anti-inflammatory hormones in the body and thereby serve to suppress the production and activity of proinflammatory cytokines during exposure to stress. Inadequate glucocorticoid-mediated feedback inhibition of the immune response will result in excess circulating levels of proinflammatory mediators. Kwon and colleagues measured the levels of proinflammatory mediators in a cohort of 82 patients, most of whom had sepsis.46 Thirty-six patients (43%) met the above cited criteria for adrenal inefficiency. The authors divided the patients with adrenal inefficiency into two groups, namely (1) those with a low basal cortisol (basal cortisol <10µg/dL) and (2) those with a basal cortisol >10µg/dL and a delta cortisol <9µg/dL. In the group of patients with a low delta cortisol the serum levels of proinflammatory mediators were markedly elevated compared to group of patients with a low basal cortisol. In the low basal cortisol group the levels of proinflammatory mediators were similar to those of the nonadrenal insufficiency control patients. These data suggest that the adrenal insufficiency subgroup with a low delta cortisol may truly have too little glucocorticoid signaling while the low baseline subgroup appears to have adequate cellular glucocorticoid activity. From a pathophysiological and therapeutic standpoint it may therefore be useful to divide adrenal inefficiency/CIRCI into two subgroups, namely, Type I, characterized by a random (stress) cortisol <10µg/dL, and Type II, characterized by a random cortisol ≥10µg/dL and a delta cortisol <9µg/dL. The practical implication of this classification is that only patients with type II CIRCI may benefit from stress doses of corticosteroids. Additional studies are required to confirm the findings of Kwon and colleagues.
TREATMENT WITH CORTICOSTEROIDS. WHO AND HOW?
Over the last three decades approximately 20 randomized controlled trials (RCTs) have been conducted evaluating the role of glucocorticoids in patients with sepsis, severe sepsis, septic shock, and ARDS. Varying doses (37.5-40,000 mg/hydrocortisone Eq/day), dosing strategies (single bolus/repeat boluses/continuous infusion/dose taper), and duration of therapy (1-32 days) were used in these studies.27,28
Related posts:






Full access? Get Clinical Tree


