KEY POINTS
Bleeding disorders and hemorrhagic complications are common in ICU patients.
Bleeding disorders may be divided into thrombocytopenias, soluble coagulation factor deficiencies, and combined disorders.
Initial management approaches to thrombocytopenias vary considerably and create the necessity for early recognition of distinct disorders including heparin-induced thrombocytopenia, thrombotic thrombocytopenic purpura, disseminated intravascular coagulation, and other common thrombocytopenias.
Disorders of soluble coagulation factors are revealed by abnormal results in the prothrombin time, activated partial thromboplastin time, and other tests including thromboelastography.
Factor deficiencies, factor inhibitors, von Willebrand disease, and other complex coagulopathies including disseminated intravascular coagulation, HELLP syndrome, massive transfusion, and anticoagulant-related syndromes have specific therapies to reduce the rate and risk of bleeding.
There are specific indications and appropriate applications for platelet transfusion, cryoprecipitate, fresh frozen plasma, concentrated and activated factors, as well as other medications, including inhibitors of fibrinolysis.
INTRODUCTION
Coagulation disorders and complications of bleeding are common and require proactive assessment and management. Intensive monitoring of ICU patients demonstrates that a substantial majority will have either a coagulation defect or bleeding. Furthermore, the coagulation abnormalities convey important prognostic information and a substantial number of patients will have severe, major bleeding.1,2 Because of the particularly high prevalence and significant impact of bleeding disorders in critically ill patients, effective and efficient ICU care requires timely recognition and mitigation of disorders of platelets, soluble coagulation factors, and vascular lesions. Appropriate management of bleeding disorders depends on recognition and adherence to specific treatment guidelines for a wide variety of patients such as those with massive transfusion and trauma, disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, and anticoagulant-related hemorrhage.
CLINICAL ASSESSMENT OF BLEEDING IN CRITICALLY ILL PATIENTS
Reliable monitoring and reporting of bleeding in critically ill patients is essential for accurate safety and performance assessments. Careful assessment of the risk and impact of hemorrhage is also critical for the appropriate selection of evidence-based treatments. However, the standards for monitoring, assessment, and treatment of ICU bleeding are highly variable. Furthermore, clinical studies utilizing bleeding assessment scales are usually constrained by application in homogenous, single-disease patients.3 The World Health Organization (WHO) took an early initiative to sponsor and develop standard grading scales for reporting complications of cancer treatment including bleeding.4 While the WHO scale (Table 90-1) is one of the most commonly reported scales, the routine use of this grading system is limited in its application in general ICU patients because it is not specifically linked to anatomic, physiologic, and therapeutic response.
ICU-based definitions of bleeding primarily rely on a dichotomy of major or minor bleeding. A practical approach is to define major bleeding as causing hypovolemic shock, affecting critical sites, requiring an invasive intervention, leading to transfusion of at least two units of red blood cells, or causing otherwise unexplained hypotension or tachycardia.5 Improved reliability of bleeding assessment results when assessment is reflective of the amount of blood loss, the rate of blood loss, and the physiologic consequence of bleeding.6 More specific criteria for major and minor bleeding that have high levels of interobserver reliability include localized anatomic criteria as well as numeric laboratory and physiologic criteria that correlate with volume of blood loss (Table 90-2).1
Categories and Criteria for Assessment of ICU Bleeding Severity1
|
FREQUENCY AND GENERAL RISK FACTORS FOR BLEEDING IN ICU PATIENTS
The risk of bleeding depends on patient and disease factors as well as specific hematologic parameters including vascular integrity, platelets and clotting factor concentrations and function. Furthermore, the reported frequency of bleeding is directly related to ascertainment issues such as the clinical definition of bleeding as well as the frequency and thoroughness of bleeding assessment. Reliable, detailed, and sensitive assessments for bleeding are typically used for therapeutic monitoring of bleeding complications in therapeutic trials involving hemostatic or anticoagulant medications. When such detailed and focused clinical bleeding evaluations were applied to a general, adult, medical-surgical ICU population of 100 consecutive patients, 90 patients were found to have had 480 discrete bleeding episodes over an average ICU length of stay of 5 days. Of these events, 95% were minor but 5% were classified as major. Risk factors for bleeding in this cohort were thrombocytopenia and prolonged coagulation times, but not renal failure or receipt of pharmacologic thromboprophylaxis.1
Using less-specific criteria, a wide range of bleeding frequencies and risk factors have been described in general, critically ill populations. Rates of bleeding as the cause of initial admission to an ICU are highly variable and depend on the medical, surgical, neurosurgical, and trauma designation of patients served by the unit. Based on descriptions of major bleeding or clinically significant bleeding, 5% to 15% of all ICU patients develop some form of bleeding after admission that has practical, clinical relevance. For example, a single-center prospective observation of 1328 patients admitted to a mixed medical-surgical ICU described 29% of patients as having bleeding present on admission, plus an additional 10% developed clinical bleeding after admission. The most common site of bleeding was gastrointestinal and the risk factors for bleeding were mechanical ventilation, malnutrition, renal failure, anticoagulant use, and antiulcer medications.7 As expected, the rate of clinically significant or major bleeding in all adult patients is lower than the subgroup of ICU patients. In large prospective study of 10,866 adult medical patients without bleeding prior to admission, the rate of bleeding after general hospital admission was observed to be 3.2%.8
Additional inferences about the risk of bleeding and bleeding complications in critically ill patients may be drawn from examination of events in disease-specific cohorts and placebo cohorts of therapeutic intervention trials. Because these observations were conducted in trial-eligible patients, caution is necessary when extrapolating to general ICU populations. The rate of clinically important gastrointestinal bleeding among a prospective cohort of adult ICU patients has been observed at 1.5% with risk factors including respiratory failure and coagulopathy.9 For patients requiring mechanical ventilation, the rate of upper gastrointestinal bleeding is higher than the general ICU population (2.8%). In medical-surgical patients eligible for heparin thromboprophylaxis, meta-analysis of placebo patients (n = 1072 patients) and patients receiving any type of heparin (n = 1084) demonstrate rates of any major bleeding of 4.9% and 4.0%, respectively.10 Similarly, in patients receiving either dalteparin or heparin thromboprophylaxis, the overall rate of bleeding was 5.6% with identified risk factors of lower platelet count, therapeutic heparin, antiplatelet agents, renal replacement therapy, and recent surgery.5 Finally, placebo cohorts of critically ill patients in severe sepsis trials demonstrate rates of clinically important or life-threatening bleeding as low as 1% to 2% and as high as 12% to 15%.11-14
PHYSIOLOGY AND MECHANISM OF NORMAL COAGULATION
Blood clot formation and maintenance of vascular patency depends on the highly complex interaction of vascular endothelium, vascular subendothelial matrix, platelets, and soluble coagulation proteins. Normal thrombus formation and subsequent dissolution result from closely regulated enzymatic activity of the coagulation factors, vascular endothelial cell surface protein expression, and recruitment and activation of platelets at the site of injury. Common clinical conditions including infection, shock, surgery, and trauma profoundly affect vascular endothelial function and coagulation pathways.15 The physiology of blood clot propagation and resolution in these clinical conditions give rise to multiple distinct clinical disorders. However, the final common result of abnormal clotting is impaired oxygen delivery to organs and tissues as the result of extravascular blood loss, altered vasoregulation, and intravascular occlusion by thrombosis.
Vascular endothelial cells and the subendothelial matrix maintain an equilibrium of coagulation activation and inhibition by directly regulating platelet adhesion, platelet activation, and the activation of soluble clotting factors. Normal vascular endothelium produces nitric oxide and prostacyclins that inhibit platelet adhesion and aggregation. During massive injury and bacterial infection, lipopolysaccharide and inflammatory cytokines mediate increases in vascular endothelial cell production of procoagulant microparticles, tissue factor, and plasminogen activator inhibitor-1, with a concomitant decrease in expression of thrombomodulin, protein C receptor, and tissue-type plasminogen activator.16-19 Vascular endothelial cell damage and mechanical trauma also expose subendothelial collagen and tissue factor, which activate both platelets and thrombin. Platelets adhere to sites of vascular injury through direct interaction of platelet receptors glycoprotein VI, Ia/IIa, and Ib-V-IX complex with the collagen and von Willebrand factor of the exposed vessel wall.20-22 Once adherent, platelets become rapidly activated and release procoagulant granules, leading to recruitment and activation of more platelets.
The central role of vascular function and integrity in critical illness is visible at the bedside in routine clinical practice. Dysfunction of the vascular endothelium resulting from atherosclerosis leads directly to pathologic thrombosis and acute coronary syndrome. Perivascular inflammation and injury in vasculitis disorders results in loss of vascular integrity, pathologic consumption of platelets, and activation of both platelet and coagulation cascades, which may be clinically visible as palpable purpura or antineutrophil antibody-associated pulmonary hemorrhage. Finally, sepsis and the systemic inflammatory response syndrome may result in disseminated intravascular coagulation and profound disruption of microvascular function causing end-organ ischemia and dysfunction.
The normal function of platelets in thrombus formation includes three overlapping events: platelet adhesion to sites of vascular injury, platelet-platelet adhesion and aggregation, and platelet activation. Platelet adhesion to exposed subendothelial collagen fibrils is mediated by interaction of platelet membrane glycoprotein receptors with subendothelial collagen matrix and circulating von Willebrand factor. Disorders of platelet glycoprotein receptors cause failure of platelet adhesion as well as failure of platelet-platelet binding and aggregation. Although rare, these adhesion diseases do result in clinical bleeding disorders such as the Bernard-Soulier syndrome (glycoprotein Ib platelet receptor deficiency). Related disorders of impaired platelet binding include von Willebrand disease which results from either quantitative or qualitative deficiency of von Willebrand factor, which is an essential cofactor in platelet-endothelial and platelet-platelet attachment.23
Platelet activation causes release of platelet granule contents and morphologic changes. Platelet activation results from platelet glycoprotein receptor attachment to collagen as well as thrombin binding to the PAR1 platelet thrombin receptor. Platelets are also strongly activated by binding circulating fibrin to integrin glycoprotein receptor IIb/IIIa, also known as integrin α(IIb)β(3) receptor. These parallel pathways of activation promote rapid and synergistic activation of platelet plug formation and coagulation cascade activation at sites of vascular injury. Platelet activation is characterized by increased platelet thromboxane A2 synthesis, protein phosphorylation, release of intracellular calcium, and subsequent changes in platelet shape and granule secretion.24 Rapid platelet degranulation releases strongly activating, procoagulant substances including adenosine diphosphate (ADP), thromboxane A2, calcium, serotonin, and platelet factor 4. These events in turn promote activation of surrounding platelets and platelet-platelet aggregation mediated by fibrin cross-linking of platelets with the IIb/IIIa receptor.25 The primary mechanisms of pharmacologic inhibition of platelet-mediated coagulation involve inhibition of platelet prostaglandin synthesis (aspirin), platelet ADP binding at the P2Y12 receptor (clopidogrel, ticlopidine, prasugrel), and inhibition of the IIB/IIIa receptor (abciximab, tirofiban, eptifibatide).
The coagulation cascade results from a stepwise activation of proenzymes to active enzymes followed by rapid amplification of coagulation activity and generation of fibrin. The coagulation cascade can be separated into discrete events that occur during normal clot formation and resolution. These steps are activation of thrombin from prothrombin, conversion of fibrinogen to fibrin, propagation and amplification of thrombin production, termination of thrombin activation, elimination of active thrombin, and enzymatic fibrin lysis (Table 90-3; Fig. 90-1). Beyond the immediate role of coagulation proteins in generation and resolution of thrombus, these enzymes have central roles in the activation and modulation of infection and inflammation.15,26
Essential Events of Coagulation Cascade
| Step | Consequence | Direct Mediator(s) | Mechanism |
|---|---|---|---|
| 1. Activation of prothrombin to thrombin | Conversion of fibrinogen to fibrin | Activated factor V | Tissue factor and calcium bind to factor VII, resulting in activation. Activated VII activates factor X, factor Xa activates prothrombin |
| 2. Conversion of fibrinogen to fibrin | Formation of insoluble fibrin multimers and cross-linking/aggregation of platelets | Thrombin | Activation of prothrombin to thrombin |
| 3. Propagation and amplification of thrombin production | Rapid burst of fibrin formation | Thrombin | Thrombin itself activates factor V to Va, factor VII to VIIIa and mediates conversion of XI to XIa |
| 4. Termination of thrombin activation and removal of active thrombin | Cessation of fibrin formation | Antithrombin, Thrombomodulin, protein C, protein S, tissue factor pathway inhibitor (TFPI) | Antithrombin neutralizes thrombin, Xa, IXa, XIIa, and XIa. Thrombomodulin binds thrombin to inhibit platelet activation and cleavage of fibrinogen. Proteins C and S inactivate factor Va and VIIIa. TFPI inhibits Xa |
| 5. Fibrinolysis | Cleavage of polymerized fibrin and release of fibrin degradation products | Plasmin, tissue-type plasminogen activator, urokinase | Plasminogen and tissue plasminogen activator form complex with fibrin leading to active proteolytic plasmin |
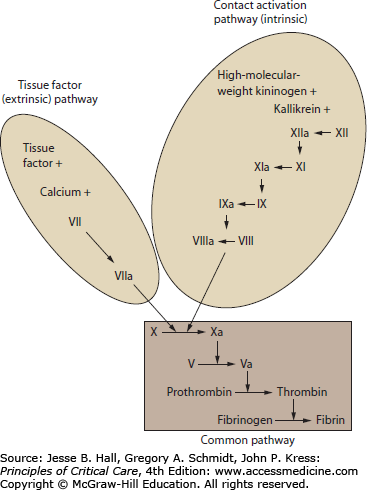
By convention, the activation of the coagulation cascade has been organized into pathways that reflect the early understanding of coagulation events and tests of coagulation function. These conceptual pathways are reflective of laboratory techniques to activate the coagulation cascade. The role of these pathways during in vivo illness may not be reflected by laboratory pathway times. In particular, the tissue factor and contact activation pathways do not account for the role of multiple-factor complexes and platelet surface interactions in clot formation. The advantage of these coagulation pathway concepts is that they facilitate understanding of common laboratory tests including prothrombin time and activated partial thromboplastin time (see “Laboratory Testing of Coagulation Function” below).27
The common pathway describes the final steps in fibrin production from activated factor X to thrombin to fibrin. The tissue factor (extrinsic) pathway is the major and most essential step in normal initiation of coagulation, beginning with calcium-dependent activation of factor VII by tissue factor.28 The contact activation (intrinsic) pathway involves activation of factors XII, XI, IX, and VIII. The contact activation pathway may not be critical for clot formation in vivo, but is believed to be involved with amplification of the cascade, fibrinolysis, vasoregulation, and modulation of inflammation.29-31 Disorders of coagulation resulting from primary disturbances of the soluble coagulation factors include congenital deficiencies (eg, hemophilia A, B), pharmacologic coagulopathies (eg, heparin, warfarin, direct factor Xa inhibitors), depletion of coagulation factors (eg, vitamin K deficiency, disseminated intravascular coagulation), severe inflammatory states (eg, sepsis, trauma), and inhibition of activity in autoimmune disease (eg, antibody inhibitors).
LABORATORY TESTING OF COAGULATION FUNCTION
The prothrombin time (PT) is an indicator of the tissue factor (extrinsic) pathway and the common pathway. Abnormally prolonged prothrombin time indicates deficiency of factor VII, prothrombin, fibrinogen, factor X, or factor V. The PT is performed by adding calcium and tissue factor to plasma then observing for clot formation by optical or electromechanical techniques as measured by seconds to clot appearance. The international normalized ratio (INR) is the ratio of the observed prothrombin time to an internationally validated control prothrombin time using a reference recombinant tissue factor activator. The main purpose of the INR is to permit valid comparison of anticoagulant effect of warfarin over time and between laboratories. While coagulation times in general do not become prolonged until at least 50% depletion or inhibition of factors, the PT typically does not become prolonged until 10% or less of normal coagulation factors are present.27 The most common causes of prolonged PT include warfarin administration, vitamin K deficiency (in poor nutrition or antibiotic-associated malabsorption), liver disease, and disseminated intravascular coagulation. Oral direct thrombin and Xa inhibitors including dabigatran, and rivaroxaban may also prolong the PT. Heparin typically does not affect the PT because the PT test reagents contain a heparin-binding chemical that eliminates its effect. Although rare, congenital deficiency of factor VII may result in a prolonged PT.
The activated partial thromboplastin time (aPTT) is an indicator of the contact activation (intrinsic) and common pathways. The test is performed by addition of a non-tissue-factor thromboplastic material (a partial thromboplastin) and negatively charged particulate contact activator (ellagic acid, kaolin, celite, or silica) to plasma. A prolonged aPTT is an indicator of deficiency of factors VII, IX, or XI in inherited hemophilia, as well as acquired states including heparin administration, lupus anticoagulant, and inhibitors of factors VII, IX, XI, or XII. In general, the factors in this pathway must decline or be inhibited to 15% to 30% of normal before the aPTT is prolonged.27
The thrombin time directly measures the time to conversion of fibrinogen to fibrin by exogenous thrombin. The thrombin time is an indicator mainly of fibrin concentration. However, dysfibrinogenemia as well as thrombin inhibitors present in the specimen may also result in prolonged thrombin time (eg, heparin, bivalirudin, argatroban, high concentrations of serum proteins in multiple myeloma). A similar test, the reptilase time (RT), uses snake venom enzyme similar to thrombin which cleaves fibrinogen but is not sensitive to heparin and thus distinguishes hypofibrinogenemia from heparin anticoagulation.
The activated clotting time (ACT) is performed by adding negatively charged particles (celite, kaolin) to a sample of freshly drawn whole blood and measuring the formation of clot. The ACT is influenced by both the soluble coagulation factors as well as the platelets present in blood sample. Although the test closely resembles the aPTT, it is clinically less sensitive to the effects of heparin and hence is used for monitoring coagulation in the setting of high heparin infusions such as coronary artery bypass surgery, percutaneous vascular procedures, and extracorporeal membrane oxygenation.
Plasmin breaks down both fibrin as well as fibrinogen into degradation products (FDP) that are recognized by fibrin degradation assays. Plasma D-dimer is only found in the degradation of cross-linked fibrin from a clot. Increases in FDP and D-dimer may indicate active fibrinolysis in patients with substantial intravascular clot burden and disseminated intravascular coagulation. Elevations in both FDP and D-dimer are common and nonspecific in ICU patients. While fibrin degradation products may be found in critical illness–related conditions such as disseminated intravascular coagulation, elevations may also be observed in nonhemorrhagic trauma, surgery, hepatic disease, pregnancy, and cancer. Importantly, patients with liver disease may have baseline elevations in FDP and D-dimer because of abnormal hepatic metabolism, but will have more substantial elevations in D-Dimer and a related protein, soluble fibrin complexes, in the setting of DIC and sepsis.32,33
Antifactor Xa activity is assayed by measuring the effect of patient plasma on a standard laboratory preparation of factor Xa. Active drugs or inhibitors in the patient serum inhibit the reaction and result in higher proportion of Xa activity. Antifactor Xa activity is used to monitor the adequacy of anticoagulation in patients receiving agents which inhibit Xa including heparin, low-molecular-weight heparin, rivaroxaban, and apixaban.
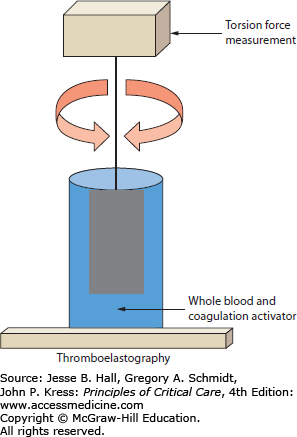
Thromboelastography is a rapid, point-of-care assay of coagulation function with both platelet and soluble coagulation factors present simultaneously. By not separating the coagulation components and by following clot formation and dissolution over time, thromboelastography may provide additional information on coagulation and fibrinolysis. The principle of measurement is based on changes in viscoelastic rotational forces between a wire or pin suspended in a cup of serum and platelets. Clot forms in the specimen cup after addition of calcium and a coagulation activator. The resulting thrombus causes adhesion and resistance to rotation which is measured electromechanically or optically in the continuously rotating system (Fig. 90-2). There are two common variations of thromboelastography that reflect commercially available equipment. In TEG (Thromboelastograph; Haemoscope/Haemonetics Corporation, United States), the torsion wire is maintained stationary while the cup containing the activated whole blood moves in a back-and-forth arc. In ROTEM (Rotation Thromboelastometry, Pentapharm GmbH, Germany), the cup is held motionless while the suspended wire is rotated.34 While conceptually comparable, these systems have different performance characteristics which prevent quantitative comparison of results between systems and which require rigorous adherence to local test standards and quality control maintenance.35
FIGURE 90-2
Thromboelastograph concept. Thromboelastography parameters are derived from measurement of the torsion forces generated by clotting blood between a suspended pin and a sample cup containing blood. As the cup or wire is rotated through an arc back and forth, clot formation increases the resistance to movement but clotting deficiencies and normal clot lysis result in decreased deflection of the pin during rotation.
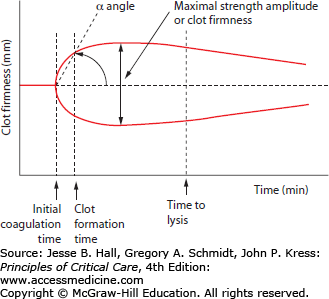
The result of thromboelastography is a graphical tracing of the change in torsion over time in the activated sample. The deflection of the cup or suspended wire is termed clot firmness which is measured in millimeters and graphed from baseline to clot formation to clot lysis over 30 to 60 minutes. There are three main components of analysis in a thromboelastograph: time, angle, and amplitude. Time-based measurements include the time to initial clotting reaction, time to 20 mm amplitude clot firmness deflection, and time to initiation of clot lysis. The angle of deflection in the clot firmness curve reflects the slope in the clotting curve and is an indicator of the velocity of initial clotting. The maximal amplitude of clot firmness indicates the overall clot strength (Fig. 90-3). The TEG and ROTEM systems use different terminology and cut-off points for these measurements which prevent direct comparison between the systems. Thromboelastography may be performed using a variety of different clot activators and inhibitors including tissue factor activators, kaolin and ellagic acid/phospholipid contact activators, heparinase/heparin degrading enzyme additives, as well as platelet activators and inhibitors. Each of these different assay variations results in different thromboelastographs and permits isolation and examination of different aspects of coagulation function, including separation of coagulation factor–based clotting behavior from platelet-based clotting.
FIGURE 90-3
Thromboelastograph measurements. The main components of thromboelastography are times to clotting events, slope of initial clot formation (α angle), and maximal strength (amplitude of deflection). The clotting event times include initial coagulation time (reaction time), clot formation time (time from initial clot formation to 20 mm deflection by definition), and clot lysis time.
Thromboelastography has been increasingly used in critically ill patients to guide blood product administration and as an indicator of severity of illness. The potential advantages of thrombelastography include point-of-care testing, ability to assess the combined result of platelet and coagulation factor activation, as well as rapid access to results. Particularly in surgeries that have typically had high utilization of blood products such as cardiac surgery, thromboelastography has been examined as a determinant of transfusion need. A randomized trial comparing intraoperative and perioperative protocols with and without thromboelastography for cardiac surgery patients showed significantly decreased blood product utilization compared to traditional coagulation times alone.36 As a prognostic indicator in general medical and surgical ICU admissions, patients with hypocoagulability on TEG assessment had higher sequential organ failure assessment (SOFA) scores, higher rate of ventilator and renal replacement therapy, and higher 30-day mortality.37 In the specific subset of medical ICU patients with sepsis and severe sepsis, thromboelastography measurements also provide prognostic information. In three separate studies involving single cohorts of sepsis patients, abnormal coagulation profiles were associated with worse SOFA scores. Patients who progressed to hypocoagulable status had significantly lower 28- or 30-day survival.38-40
PLATELET DISORDERS
Coagulopathy in critically ill patients may result from disorders of platelet number as well as platelet function. Abnormal circulating platelet number results from increased destruction, decreased synthesis, sequestration from the circulation, and combinations of these problems. Disorders of platelet function arise from impaired or inhibited platelet adhesion, activation, and degranulation. Common findings in patients with clinically significant thrombocytopenia include petechial hemorrhages and ecchymoses when thrombocytopenia is mild to moderate. When more severe, florid purpura and bleeding from the nose gums, gut, urinary tract, intravenous access sites, and surgical wounds are seen. Independent of etiology, worsening thrombocytopenia and inhibition of normal platelet function are associated with increased ICU morbidity and mortality as well as increased utilization of resources. While some defects in platelet number and function may improve with platelet transfusion, specific platelet transfusion guidelines limit the use of this treatment to actively bleeding patients and to those who have extreme thrombocytopenia plus risk factors for hemorrhage.
The life span of platelets in the circulation is limited to 9 to 10 days.41 In the absence of disease, platelet life span is likely related to the attenuation of proteasome function in the platelets themselves which then triggers conformational changes in surface protein expression and clearance from the circulation through phagocytosis by macrophages.42 Given this rapid turnover, any disorder which has a negative effect on platelet production, circulation, or clearance can quickly lead to a cumulative result of clinically significant thrombocytopenia.
Thrombocytopenia is one of the most common coagulation abnormalities critically ill patients. The reported incidence and prevalence of thrombocytopenia varies by the clinical definition applied and with subgroups of ICU patients.43 Applied general definitions of thrombocytopenia are platelet concentrations below 150 × 109/L or below 100 × 109/L. A small number of reports use cut-offs of 50 × 109/L to specifically designate severe thrombocytopenia. Typical definitions are mild thrombocytopenia—concentrations of 100 to 149 × 109/L; moderate—50 to 99 × 109/L; and severe—<50 × 109/L.44 Using a cut-off of 150 × 109/L or lower, the prevalence of thrombocytopenia in medical, surgical, and trauma patients on admission to the ICU shows a wide range from 8.3% to 68%.45 Following admission, the incidence of new thrombocytopenia ranges from 13% to 44% of patients.45,46 Severe thrombocytopenia (<50 × 109/L) is significantly less common and is described in 1.6% to 15% patients.44,47
Thrombocytopenia is associated with multiple adverse outcomes and functions as an indicator of disease severity. Adjusted mortality rates and severity of illness scores are generally higher in patients with worsening severity of thrombocytopenia, lower nadir platelet counts, or a relative drop in platelet count of 30% or more.44-48 Thrombocytopenia has been associated with use of inotropes and vasopressors, renal replacement therapy, and liver dysfunction.44 Thrombocytopenia has also been associated with increased rates and duration of mechanical ventilation and blood product utilization.46,47 While disease-specific cohort studies have shown a relationship of bleeding and hemorrhage to severe thrombocytopenia, this generalization has not been consistently observed in unselected ICU patients. A systematic review found little evidence linking generally defined thrombocytopenia with hemorrhage.45 This suggests that additional factors including the etiology of thrombocytopenia, degree of thrombocytopenia, comorbid conditions, and other concurrent coagulation defects contribute to bleeding risk. Patients with severe thrombocytopenia (platelet count <20 × 109/L) have the highest risk and incidence of bleeding.49
The majority of patients who develop thrombocytopenia in the ICU develop this by day 3 to 5. Those who recover and survive demonstrate a more rapid and complete recovery of platelet count compared to nonsurvivors who have a delayed recovery in platelet counts or no recovery.50,51 Persistent absolute thrombocytopenia in patients on ICU day 7 or a relative decrease in platelet counts of 30% from baseline independently predicts higher risk of death.48 These findings of delayed recovery of platelet counts even in patients without absolute thrombocytopenia suggest that changes over time are particularly important and may be more helpful indicators of patient status than any single measurement of platelet concentration.
Identification of the causes of thrombocytopenia requires a systematic search for disorders of platelet production, destruction, dilution, and sequestration. Particularly for critically ill patients, the greatest likelihood is that multiple, simultaneous factors contribute to thrombocytopenia. Severe thrombocytopenia should be investigated by review of nutritional factors, medications, autoimmunity diseases, infection, coagulation, and bone marrow function. Important coexisting defects in soluble coagulation factors as well as comorbid conditions should be used to determine both the risk associated with thrombocytopenia and appropriate therapy. The approach to determining the cause of thrombocytopenia should include assessment of the patient’s history, exposures, and medications, as well as consideration of risk for malignancy and disseminated infection. Laboratory testing should include review of the complete blood count, prothrombin time, nutritional markers, and hepatic function. While routine bone marrow biopsy has been shown to add important new information to the understanding of thrombocytopenia, this should be reserved for patients with refractory, unexplained disease.
A highly detailed evaluation of 301 mixed adult ICU patients which included bone marrow biopsy, demonstrated that the majority of patients had at least two, but commonly three or four, potential etiologies for thrombocytopenia. In this cohort, the most important causes of thrombocytopenia were sepsis, disseminated intravascular coagulation, dilutional thrombocytopenia, massive transfusion, drug-induced, folate deficiency, and malignancy. 49Table 90-4 lists the most common etiologies and clinical characteristics of thrombocytopenia in ICU patients.
Etiologies of Thrombocytopenia
| Spurious |
|
|
|
Spurious Thrombocytopenia and Pseudothrombocytopenia: The first assessment in any patient with thrombocytopenia is to confirm that the result is consistent with the clinical scenario and that there were no errors in measurement. Routine platelet concentration measurement is performed by an automated analyzer that is triggered by the relative size of the particles and cells in the sample. Errors in sample collection including incorrectly labeled specimens, delays in processing, and inadequate anticoagulant in the collection tubes may allow platelets to clump and cause the automated counter to generate incorrect or falsely low results. Ethylenediaminetetraacetic acid (EDTA) anticoagulant used in peripheral blood collection tubes may also trigger antibody-dependent clumping or agglutination of platelets leading to low measured platelets in patients with severe illness, autoimmune, liver, and neoplastic diseases.52 In these instances of pseudothrombocytopenia, examination of the peripheral blood by microscopy quickly reveals the actual platelet density as well as other features of the problem such as giant or clumped platelets. While manual recounting of platelets on peripheral blood smear may be performed, it is recommended that patients with clumped platelets have repeat testing using citrate anticoagulant or heparin anticoagulant specimen tubes as this will facilitate continued monitoring and assessment of actual platelet count over time.
Decreased Platelet Production and Disorders of Bone Marrow: The short, limited life span of platelets creates a physiologic requirement for rapid platelet production to maintain steady-state circulating levels. Nutritional deficiencies, toxins, and infectious diseases which affect bone marrow stem cells, specifically megakaryocytes, can cause thrombocytopenia. The most common causes of decreased platelet production in ICU patients include hematologic malignancies, nonhematologic malignancies, aplasia after chemotherapy, vitamin B12 and folate deficiencies, and bacterial and viral infections.49 While iron deficiency is typically associated with thrombocytosis, it may also be associated with thrombocytopenia.53 Clinical features that suggest impaired bone marrow production include specific medical history (eg, cancer, alcohol ingestion, medications, dietary habits, chemotherapy, or radiation), and concurrent red blood cell and white blood cell abnormalities (eg, leukopenia, microcytic, or macrocytic anemia). If suspected, specific viral infections including HIV, Epstein-Barr virus, parvovirus, and varicella may be diagnosed by history and serologic testing.
Dilution and Distributional Thrombocytopenia: Dilutional thrombocytopenia reflects inadequate preservation of platelet concentration when intravascular volume is rapidly replaced with platelet-poor solutions and crystalloid. For practical purposes, this only occurs in settings of massive blood loss and massive transfusion (see massive transfusion below). Distributional thrombocytopenia occurs in patients with splenomegaly from portal hypertension and reflects removal and sequestration of platelets from the circulation. However, patients with advanced portal hypertension and splenomegaly are also likely to have decreased thrombopoietin production associated with cirrhotic liver disease and may have alcohol-related bone marrow suppression.54,55
Drug and Medication-Induced Thrombocytopenia: Hundreds of drugs, supplements, and foods have been causally associated with thrombocytopenia. While specialized laboratory testing may be available to confirm the diagnosis of drug and medication-induced thrombocytopenia (DITP), the clinical diagnosis is often established only by clinical consideration of the time course, response to removing the drug, and exclusion of alternative causes. These factors require a high index of suspicion and a systematic review of potential drug-related etiology in any patient with thrombocytopenia. The most typical clinical feature of DITP is clinical presentation 5 to 10 days after exposure to the drug with platelet counts usually less than 20 × 109/L.56 Clinical criteria and a scoring scale for the likelihood of DITP have been proposed. One commonly applied scale assigns probability based only on clinical information of exposure and response to removal of exposure and does not require specialized laboratory testing evidence (Table 90-5).57
Criteria and Clinical Likelihood for Drug-Induced Thrombocytopenia
| Criterion | |
| 1 | Therapy with the candidate drug preceded thrombocytopenia and recovery from thrombocytopenia was complete and sustained after therapy with the drug was discontinued |
| 2 | The candidate drug was the only drug used before the onset of thrombocytopenia or other drugs were continued or reintroduced after discontinuation of therapy with the candidate drug with sustained normal platelet count |
| 3 | Other causes for thrombocytopenia were excluded |
| 4 | Reexposure to the candidate drug resulted in recurrent thrombocytopenia |
| Clinical Likelihood | |
| Definite | Criteria 1, 2, 3, and 4 met |
| Probable | Criteria 1, 2, and 3 met |
| Possible | Criterion 1 met |
| Unlikely | Criterion 1 not met |
The most common specific classes of drugs associated with idiosyncratic or unpredictable thrombocytopenia include the cinchona alkaloids, platelet inhibitors, heparins, antibiotics, anticonvulsants, and analgesics. Thrombocytopenia is anticipated and predictable with cytotoxic chemotherapeutic and immunosuppressive medications. Common medications associated with DITP are shown in Table 90-6. Specific medications strongly associated with DITP and consistently detected antiplatelet antibodies include quinine, quinidine, trimethoprim/sulfamethoxazole, vancomycin, penicillin, rifampin, carbamazepine, ceftriaxone, ibuprofen, mirtazapine, oxaliplatin and suramin, abciximab, tirofiban, eptifibatide, and heparin.58 Importantly, antibody-mediated platelet destruction may result not only in removal of circulating platelets and bone marrow megakaryocytes, but also may cause significant reductions in the productive capacity of surviving megakaryocytes.59
Common Drugs Associated With Thrombocytopenia
| Drug Class | Specific Drugs |
|---|---|
| Antibiotics | Penicillin, methicillin, vancomycin, rifampin, ciprofloxacin, trimethoprim/sulfamethoxazole and sulfonamides, linezolid, rifampin, amphotericin B |
| Analgesics | Ibuprofen, diclofenac, naproxen, acetaminophen |
| Cinchona alkaloids | Quinine, quinidine |
| Cardiac glycosides and antiarrhythmics | Digoxin, procainamide, amiodarone |
| Chemotherapeutic and immunosuppressive agents | Oxaliplatin, fludarabine, cyclosporine, rituximab, gold salts, d-penicillamine |
| Diuretics | Chlorothiazide, hydrochlorothiazide |
| Heparins | Unfractionated heparin, low-molecular-weight heparin |
| Histamine-receptor antagonists | Cimetidine, ranitidine |
| Platelet inhibitors | Abciximab, eptifibatide, tirofiban |
| Sedatives and antiseizure agents | Carbamazepine, haloperidol, phenytoin, valproic acid, diazepam |
Heparin-Induced Thrombocytopenia: Heparins, including unfractionated heparin and, to a lesser degree, low-molecular-weight heparin (LMWH), are among the most common drugs associated with thrombocytopenia. Heparin-induced thrombocytopenia (HIT) results from immune-mediated activation and destruction of platelets. Specifically, HIT is caused by induction of a specific immune antibody response which results in immunoglobulin binding to FcγIIa receptors on platelets and monocytes.60,61
Related posts:






Full access? Get Clinical Tree


