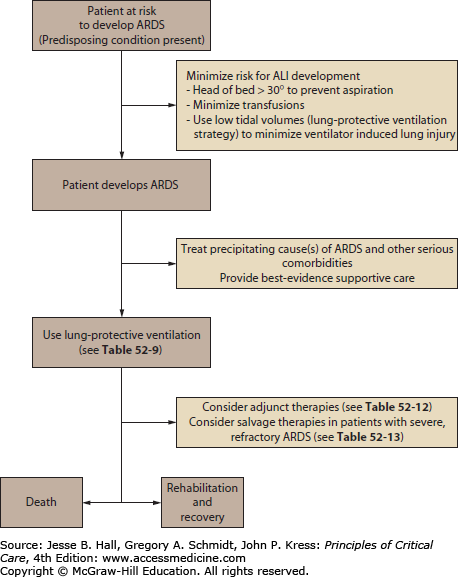
KEY POINTS
Acute lung injury (ALI) and its more severe form, the acute respiratory distress syndrome (ARDS), are common causes of acute hypoxemic respiratory failure (AHRF). The 2012 Berlin Definition eliminated the term ALI; however, this term remains common to older literature.
Both ALI and ARDS are characterized by hypoxemia that is resistant to oxygen therapy; this is due to widespread alveolar filling or collapse.
Initial therapy for all patients with ALI and ARDS should be supplemental oxygen; failure to achieve 95% arterial saturation or greater confirms the presence of a large right-to-left shunt.
Most patients with ALI and ARDS require ventilatory support because their AHRF is typically severe and may be prolonged.
If a patient with severe hypoxemia as indicated by arterial blood gas analysis has a clear chest radiograph, consider a possible error (eg, incorrect fractional inspired oxygen [FiO2] or arterial oxygen tension [PaO2]); in such situations, also consider the possibility of other types of right-to-left shunts (eg, intracardiac shunts or pulmonary arteriovenous malformations) or the continued perfusion of an unventilated or poorly ventilated lung (eg, due to acute mucous plugging of one main bronchus).
The acute phase of ALI and ARDS is characterized by an exudative alveolar flooding due to pulmonary capillary leak and by extensive alveolar collapse due to loss of normal surfactant activity; while interventions directed at modulating inflammatory or other pathways of lung injury, optimizing alveolar fluid clearance, or restoring surfactant function hold theoretical promise, at present no specific pharmacologic therapy has been shown to improve outcomes; currently one should provide lung-protective mechanical ventilation and other supportive care while identifying and treating the precipitating causes of ALI or ARDS.
Lung-protective ventilation of patients with ALI and ARDS should use a strategy with low tidal volumes and limits to end-inspiratory pressure (ie, plateau pressures [Pplat]), to reduce the risk of ventilator-induced lung injury (VILI); such a strategy gives higher priority to the goal of decreasing the risk of VILI by limiting end-inspiratory lung volume and pressure than the traditional goal of keeping arterial carbon dioxide tension (PCO2) and pH in the normal range.
The target for oxygenation should be a PaO2 between 55 and 80 mm Hg (88%-95% saturation); one should achieve this by adjusting FiO2 and positive end-expiratory pressure (PEEP) with the goal of decreasing FiO2 to 0.5 to 0.6 (or less), concentrations that are less concerning for pulmonary oxygen toxicity. However, despite this recommendation, it should be noted that the safety of a permissive hypoxemia approach on nonpulmonary organ function has not been demonstrated.
In general, the ventilatory strategy should start with a tidal volume of 6 mL/kg of predicted body weight (PBW) and with a Pplat target that does not exceed 30 cm H2O; if Pplat exceeds 30 cm H2O with a 6 mL/kg PBW tidal volume, the latter should be decreased to 5 mL/kg PBW; if Pplat still exceeds 30 cm H2O, tidal volume should be further decreased to 4 mL/kg PBW.
When changing to low-tidal-volume ventilation, one should increase the respiratory rate (RR) up to 35/min to maintain minute ventilation; the target for ventilation should be a pH of 7.30 to 7.45; if the pH is between 7.15 and 7.29, if the pH remains <7.15 with RR of 35/min and NaHCO3 is considered or given, one can increase the tidal volume in 1-mL/kg PBW increments until pH exceeds 7.15 (ie, the Pplat target may be exceeded).
The use of a recruitment maneuver (eg, 30 cm H2O for 30 seconds or 40 cm H2O for 40 seconds), coupled with an increase in positive end-expiratory pressure (PEEP) to maintain the recruitment achieved, should be considered in patients with refractory hypoxemia; however, clinicians should be vigilant for the development of barotrauma or hypotension as a consequence of the maneuver, and the trial should be ceased if either complication develops.
During assisted ventilation, one should use analgesia and/or sedation at the lowest doses required to synchronize the patient’s respiratory efforts with the ventilator and to decrease oxygen consumption (˙VO2); one may also need to induce muscle paralysis by use of neuromuscular blocking agents (NMBA) and recent evidence suggests that early NMBA use in severe ARDS may be associated with improved outcomes.
The use of a “dry” fluid management strategy should be considered given evidence that this approach improves lung function and shortens duration of mechanical ventilation and intensive care unit stays without increased nonpulmonary organ dysfunction in the short term; however, this approach generally lowers cardiac output and the question has been raised if its use may be associated with long-term cognitive impairment.
If one uses a “dry” fluid management strategy, one should be guided by daily and cumulative net intake and output volumes and laboratory indices of hypovolemia (blood urea nitrogen [BUN]:creatinine ratio and serum total bicarbonate) while monitoring for adequate urine output (>0.5 mL/kg PBW/h), effective circulation (by physical signs), or invasive measurements (ie, mixed [or superior vena caval] venous cooximetry), adequate perfusing pressure (ie, mean arterial pressure [MAP] ≥60 mm Hg), and electrolyte abnormalities (serum sodium and potassium).
The use of a pulmonary artery catheter (PAC) to guide therapy, compared to a central venous catheter (CVC), is associated with more complications and increased costs without a demonstrable benefit in outcomes; the PAC is not recommended for routine use in the management of patients with ALI and ARDS.
Late-phase or fibroproliferative-phase ARDS (corresponding to persistent ARDS of 1-week duration or more) is characterized by subacute inflammation, proliferation of alveolar lining cells and interstitial cells, and varying degrees of fibrosis.
In severe late-phase ARDS, a prolonged course of high-dose methylprednisolone sodium succinate (MPSS) can improve gas exchange and mechanics in some patients; however, use of MPSS increases the risk of diffuse weakness that may be prolonged; results from one RCT found that MPSS administration when added to a low-tidal-volume ventilation strategy that limited Pplat as described above did not significantly improve mortality at 60 days (or ICU or hospital lengths of stay).
In refractory ARDS, the use of extracorporeal life support at experienced centers, as a means to temporarily support cardiopulmonary function and minimize VILI and oxygen toxicity, may improve survival.
Severe arterial hypoxemia that is resistant to supplemental oxygen is a common reason for admission to the ICU. This form of respiratory failure, termed acute hypoxemic respiratory failure (AHRF), arises from widespread flooding or collapse of alveoli. As a result, a substantial fraction of mixed venous blood traverses nonventilated alveoli (ie, alveoli with ventilation-perfusion ratio of 0). This in turn results in a large right-to-left intrapulmonary shunt (Fig. 52-1). In addition to its adverse effects on oxygenation, fluid that accumulates in alveoli and interstitial tissues increases lung stiffness, which decreases lung compliance. This imposes a larger mechanical load on the patient’s respiratory system, resulting in an increased elastic work of breathing. Without treatment with supplementary oxygen and assisted ventilation, the gas exchange derangement and abnormal lung mechanics can result in progressive hypoxemia, respiratory muscle fatigue, and eventual respiratory arrest and death.
FIGURE 52-1
Diagram of a two-compartment model of lung perfusion and ventilation demonstrating the basis for failure of oxygenation in ALI and ARDS. When large portions of the lung are nonventilated owing to alveolar collapse or flooding (hatched area), blood flow to these units with mixed venous  (
(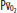 )(
)(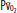 ) of 40 mm Hg and content of 15 vol% is effectively shunted through the lungs without being resaturated. Thus despite a high concentration of supplemental oxygen (100% in this example) and a high alveolar
) of 40 mm Hg and content of 15 vol% is effectively shunted through the lungs without being resaturated. Thus despite a high concentration of supplemental oxygen (100% in this example) and a high alveolar  in ventilated units, these blood flows mix in accord with their oxygen contents (ie, the resulting left atrial blood has an oxygen content that is the weighted mean of the oxygen content of the shunted and non-shunted blood). In this example of a 50% shunt, the left atrial and systemic arteries have an arterial
in ventilated units, these blood flows mix in accord with their oxygen contents (ie, the resulting left atrial blood has an oxygen content that is the weighted mean of the oxygen content of the shunted and non-shunted blood). In this example of a 50% shunt, the left atrial and systemic arteries have an arterial 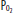 of 60 mm Hg.
of 60 mm Hg. 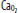 , arterial oxygen content; CO2, capillary oxygen content;
, arterial oxygen content; CO2, capillary oxygen content; 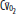 , mixed venous oxygen content; Pa, alveolar pressure;
, mixed venous oxygen content; Pa, alveolar pressure; 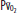 , arterial oxygen pressure;
, arterial oxygen pressure; 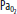 , partial pressure of oxygen in the mixed venous blood.
, partial pressure of oxygen in the mixed venous blood.
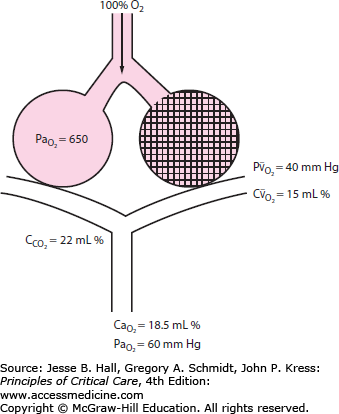
This chapter describes two traditional notable forms of AHRF: acute lung injury (ALI) and its more severe subgroup, the acute respiratory distress syndrome (ARDS) (Fig. 52-2). Though the term ALI has been eliminated in the 2012 Berlin definition of ARDS, its use is seen frequently in older literature (see below). ALI and ARDS affect up to an estimated 190,000 patients each year in the United States alone.1,2 Despite recent advances in ventilatory management and improvements in nonrespiratory supportive ICU care, the mortality rate remains high (eg, ∼25% to over 50%, depending on the characteristics of the population studied and the ventilatory strategy utilized).2-9
FIGURE 52-2
Schematic representation of the traditional relationships among acute hypoxemic respiratory failure (AHRF), acute lung injury (ALI), and the acute respiratory distress syndrome (ARDS). Note that ALI is a more severe subgroup of AHRF and that ARDS is a more severe form of ALI. The more recent Berlin definition of ARDS categorizes ARDS as mild, moderate, and severe.24
The overall goal of this chapter is to provide a comprehensive and updated clinical approach to ALI and ARDS that is evidence-based and grounded in current concepts of pathogenesis and pathophysiology. Objectives include (1) to present the definitions, epidemiology, and precipitating causes of ALI and ARDS; (2) to describe efforts to predict the development of ALI and ARDS and to prevent its development; (3) to describe the pathogenesis and pathophysiology of ALI and ARDS; (4) to describe the differential diagnosis of ALI and ARDS and clinical approaches to distinguish among various causes of AHRF; (5) to review the current status of treatment for ALI and ARDS and their precipitating causes; (6) to provide current recommendations for ventilatory management of ALI and ARDS; (7) to review so-called “salvage” therapies for refractory ARDS. The last objective includes a detailed description of the low-tidal-volume ventilatory strategy used by the ARDS Clinical Trials Network (ARDSNet) investigators in the clinical trials sponsored by the National Heart, Lung, and Blood Institute (NHLBI) of the National Institutes of Health (NIH).3,4
DESCRIPTION AND DEFINITIONS OF ALI AND ARDS
ARDS was initially described in 1967 by Ashbaugh and coauthors as a syndrome characterized by the acute onset of dyspnea, severe hypoxemia, diffuse lung infiltrates, and decreased compliance of the respiratory system.10 Following this initial report, different authors utilized varying definitions that incorporated the elements of acute timing of onset, hypoxemia, radiographic infiltrates, and absence of overt clinical congestive heart failure.11-15 In 1988, Murray and coauthors proposed using a Lung Injury Score (LIS), in part to take into account the effects of respiratory system compliance and the pressures delivered using the mechanical ventilator.14 The LIS combined elements of severity of chest radiograph infiltrates, hypoxemia, respiratory system compliance, and amount of administered positive end-expiratory pressure (PEEP) on the mechanical ventilator.14
In 1994, the first American European Consensus Conference (AECC) published these consensus-derived definitions of ALI and ARDS: acute onset, bilateral pulmonary infiltrates on chest radiograph consistent with pulmonary edema, poor systemic oxygenation, and the absence of evidence of left atrial hypertension as a means to discriminate permeability pulmonary edema from predominantly hydrostatic pulmonary edema (Table 52-1).16 The ratio of arterial oxygen tension (PaO2) to the fraction of inspired oxygen (FiO2), PaO2 : FiO2, was chosen to reflect the degree of hypoxemia even when measured at different FiO2.17 The syndrome was named ALI when this ratio is ≤300 and ARDS when ≤200. The AECC coined the term acute lung injury in order to identify patients who are early in the course of their ARDS and those who may have a form of AHRF that is milder than ARDS. The AECC definitions of ALI and ARDS were intentionally broad in order to encompass different types of AHRF occurring in a wide variety of settings. Most patients with ALI progress to ARDS, prompting some to use the term “ALI/ARDS” to describe all patients with a PaO2 : FiO2 ratio ≤300 who also meet the other AECC criteria (see Table 52-1).
American European Consensus Conference Criteria for Acute Lung Injury (ALI) and the Acute Respiratory Distress Syndrome (ARDS)
| Clinical Variable | Criteria for ALI | Criteria for ARDS |
|---|---|---|
| Onset | Acute | Acute |
| Hypoxemia | PaO2/FiO2 ≤300 mm Hg | PaO2/FiO2 ≤200 mm Hg |
| Chest radiograph | Bilateral infiltrates consistent with pulmonary edema | Bilateral infiltrates consistent with pulmonary edema |
| Noncardiac cause | No clinical evidence of left atrial hypertension or, if measured, pulmonary artery occlusion pressure ≤18 mm Hg | No clinical evidence of left atrial hypertension or, if measured, pulmonary artery occlusion pressure ≤18 mm Hg |
Several clinical trials have used these standardized definitions of ALI and ARDS to specify the trial’s inclusion criteria for their study populations.3,4,8 Using the AECC definition of ALI and ARDS in these and other clinical trials contributes to the comparability of their study populations and facilitates generalizability of their results. For example, in order for clinicians to extrapolate the results of a trial to their own patients, they must first be reasonably sure that their patients resemble those in the clinical trial. This requires that they meet the same inclusion criteria for ALI or ARDS as those used in the clinical trial. However, having the same inclusion criteria is necessary but not sufficient, since the clinician must also take into account the exclusion criteria used in the clinical trial to assess comparability.
Despite standardization, these definitions have not been formally validated beyond their face validity. In addition, problems remain with the reliability of the various components of the definitions.1 For example, interpretation of chest radiographs can be inaccurate and variable among different observers.18,19 However, formal training sessions can improve this variability.19 Likewise, the PaO2 : FiO2 criterion also suffers from variability since it can be influenced by the level of PEEP used in mechanical ventilation and other transient factors, such as airway secretions or inadequate sedation. For example, higher PEEP generally increases PaO2 at a given FiO2. This in turn may raise a patient’s PaO2 : FiO2 >300 so that the patient no longer meets the inclusion criteria for ALI. Conversely, in the absence of PEEP a PaO2 : FiO2 <300 may reflect the presence of simple basilar atelectasis rather than ALI or ARDS. In such a case, adding PEEP may recruit enough atelectatic lung to raise PaO2 : FiO2 >300, so that the patient no longer meets this criterion for ALI.20
Finally, exclusion of congestive heart failure (left atrial hypertension) also presents problems with reliability. Diagnostic criteria for left atrial hypertension on purely clinical grounds can be inaccurate.1,21 Insertion of a pulmonary artery catheter for this purpose may also be inconclusive since the pulmonary capillary wedge pressure (Ppw) may be higher than 18 mm Hg due to intravascular volume loading (eg, in patients with shock), rather than due to congestive heart failure.21 In a recent clinical trial, 29% of the ALI subjects enrolled had a Ppw in excess of 18 mm Hg; importantly, only 3% of these subjects had a depressed cardiac index.22 Conversely, many patients with hydrostatic pulmonary edema due to congestive heart failure and high left atrial pressures may have normal pulmonary artery occlusion pressures by the time the catheter is inserted and measurements taken.23 Together, the evidence demonstrates that a hydrostatic pressure gradient is present in many ALI patients, which contributes to the development of pulmonary edema.
The need for further refinement of the reliability and validity of definitions of ALI and ARDS prompted an international expert panel endorsed by multiple societies to convene in 2011 to revise the definition. The results of the revised, consensus draft definition for ARDS, known as “The Berlin Definition,” were the following: (1) the radiographic elements of the AECC definition were retained, (2) the timing of ARDS was clarified to occur within 1 week of an acute insult, (3) the Ppw criteria was removed and, acknowledging that left atrial hypertension may coexist with ARDS, it was stipulated that the etiology of pulmonary edema not be due to hydrostatic pulmonary edema exclusively, and (4) the terminology ALI was removed and ARDS was categorized into one of three mutually exclusive categories of hypoxemia severity, being mild (200 < PaO2 : FiO2 ≤ 300 with PEEP at a minimum of 5), moderate (100 < PaO2 : FiO2 ≤ 200), and severe (PaO2 : FiO2 ≤ 100).24 In the remaining chapter, we use the traditional ALI and ARDS terminology as the evidence which is presented is based predominantly on the AECC criteria.
EPIDEMIOLOGY
Inconsistent definitions for ALI and ARDS in large databases of diagnoses for hospital admissions or complications, variations in the application of the AECC definition in different studies, and variations in different at-risk populations (such as sepsis, trauma, surgeries, and bone marrow transplants) previously hampered obtaining accurate estimates of the incidence of ALI and ARDS. More recent studies have used data from the ARDSNet clinical trials and regional cohort studies to arrive at estimates in the United States in the range of 25 to 79 cases per 100,000 person-years.1,2,25,26 A recent epidemiologic study based out of Olmstead County, Minnesota, found that ARDS incidence decreased from 82 to 39 per 100,000 person-years between 2001 and 2008.26 The authors hypothesized that improvements in health care delivery (eg, use of lower tidal volumes and less blood transfusions in at-risk patients) contributed to the reduced incidence26 and efforts are underway to determine whether a bundle of ALI prevention strategies can reduce the risk of ALI development in at-risk patients.
Based on these estimates of incidence, the number of patients with ALI and ARDS in the United States each year is estimated to be between 53,000 and 190,000. If one assumes a short-term mortality of ∼30% to 40%, the annual number of deaths in the United States directly attributable to ALI and ARDS is between 16,000 and 74,500. These estimates of attributable mortality are greater than from the acquired immune deficiency syndrome (AIDS), asthma, or cervical cancer.1,2
These estimates also highlight that more than 100,000 patients will survive ALI each year. As described in Chap. 15, many of these ALI survivors will suffer from long-term physical and neuropsychological impairment, decreased quality of life, and their long-term mortality is affected.27-31 Collectively, the incidence, case-fatality, and long-term consequences demonstrate the important public health impact of ALI and ARDS.
ALI and ARDS can be considered to be a “final common pathway” reaction of the lung to a large variety of precipitating causes. Some authors have classified these causes as representing direct (pulmonary) or indirect (extrapulmonary or systemic) injury to the lung32,33 (Table 52-2). Although the methodology for applying these labels is not standardized and thus may be inconsistent between studies, this construct may have pathogenetic and pathophysiologic underpinnings since indirect causes may have different mechanisms of injury to the lung compared to direct causes.34
Examples of Direct and Indirect Precipitating Causes of Acute Lung Injury (ALI) and the Acute Respiratory Distress Syndrome (ARDS)
| Direct Precipitating Cause | Indirect Precipitating Causea |
|---|---|
| Aspiration of gastric contents | Acute pancreatitis |
| Bacterial pneumonia (eg, Legionnaire disease) | Blood product transfusions with transfusion-related acute lung injury (TRALI) |
| Chest trauma with lung contusion | Post-cardiopulmonary bypass |
| Near-drowning | Primary graft failure of lung transplantation |
| Pneumonia due to Pneumocystis jiroveci | Severe sepsis and septic shock |
| Toxic inhalations (eg, smoke inhalation, inhaled crack cocaine [“crack” lung]) | Toxic ingestions (eg, aspirin, tricyclic antidepressants) |
| Viral pneumonia (eg, influenza, the severe acute respiratory syndrome [SARS]) | Trauma with multiple fractures and fat-emboli syndrome |
Not all patients with these precipitating conditions develop ALI/ARDS. Indeed, the frequency of ALI/ARDS is quite variable. Depending on the precipitating cause, and the population studied, it ranges from ∼5% to 40%.32-37 If patients have more than one of these precipitating causes, they are at an even higher increased risk for development of ALI or ARDS. On average, ALI develops early in the course of the hospitalization in at-risk patients. In two, large multicenter observational cohort studies, ALI developed on average by hospital day 2 and the vast majority within 4 days.35,36 Patients with a direct injury to the lung developed ALI on average 3 days earlier than patients with an indirect injury.35
Why certain individuals with the same precipitating cause develop ALI and ARDS while others do not is unknown. Some of this differential risk can be attributed to other acquired risk factors, such as chronic alcoholism.37 In addition, certain individuals may have an inherent predisposition to developing ALI or ARDS while others may have inherent protections against ALI or ARDS. These considerations are discussed below.
ALI and ARDS arise from diverse populations. Within these populations, specific clinical variables may affect both the risk of ARDS and ultimate outcomes if ARDS develops.34-36,38,39 Clinical variables and outcomes associated with an increased risk of ARDS include chronic alcohol abuse37; lack of diabetes40; hypoproteinemia41; age and gender13; severity of injury and illness as measured by injury severity score (ISS) or Acute Physiology and Chronic Health Evaluation (APACHE) score13; and possibly cigarette smoking.42 The mechanistic underpinnings of these associations are the subject of ongoing research. Furthermore, processes of care, including transfusion of blood products13,43 and initial ventilator settings (eg, tidal volumes greater than 6.5 mL/kg PBW),44,45 have been associated with an increased risk of ALI development. These observations highlight the influence of the heterogeneity of a diverse source population and diverse processes that impact on the development of ALI and ARDS.
Based on the identified predisposing conditions and risk modifiers for the development of ALI, a recent study validated a Lung Injury Prediction Score (LIPS) to identify patients at high risk for the development of ALI.36 The LIPS has the potential to be used clinically to identify at-risk patients and prevention strategies directed at the at-risk population identified are warranted.
Human studies aimed at investigating genetic or molecular mechanisms that predispose or protect individuals from developing ALI and ARDS must take into account the effects of these population and environmental differences. The initial investigations into genetic determinants of ALI have been promising, including the first successful genome-wide association study, which identified a genetic variant that demonstrates the role of cell-matrix interactions in ALI development,46 yet as detailed in several recent reviews, few associations have been replicated.47-49 To date, the novel genetic variants that appear to alter ALI risk relate to inflammation and the immune response, the endothelium, the epithelium, and lipid metabolism.49-54
Clinical variables associated with increased mortality among patients with ALI and ARDS include advanced age, gender, race, low PaO2 : FiO2, high plateau pressure (ie, low compliance), extent of pulmonary infiltrates, chronic liver disease, nonpulmonary organ dysfunction, sepsis, severity of illness, hypoproteinemia, and length of hospitalization prior to ARDS.4,55-61 In addition, magnitude of the dead space fraction has been identified as a risk factor for mortality, possibly indicating the importance of loss of the pulmonary vascular bed in disease severity.62
Different precipitating causes of ALI and ARDS carry different prognoses. For example, trauma-associated ALI or ARDS has a better prognosis than other causes, even after adjustment is made for other baseline variables such as age.57 Sepsis-associated ALI, due in part to the severity of illness at presentation and coexisting comorbidities, has a worse prognosis.58 Although response to some therapeutic interventions in ALI or ARDS varies according to direct or indirect cause of lung injury,33,63 low-tidal-volume ventilation is equally efficacious in both of these subgroups.64
Despite recent advances in therapy, the mortality rates reported in clinical trials in the past decade have ranged from ∼25% to more than 50%.3-9,22 The lowest mortality rates have been achieved with low-tidal-volume ventilation, which also limited end-inspiratory (plateau) pressure.3,4,22 Although no recent studies have explicitly examined the cause of death in nonsurvivors, previously the majority of deaths were found to be due to multiorgan failure or sepsis, while progressive respiratory failure accounted for a small minority of deaths (eg, ∼15%).65,66
ALI and ARDS impact the health and lives of survivors beyond the ICU. Long-term physical, neuropsychiatric, and cognitive impairments in ALI survivors are common and are associated with reduced quality of life (see Chap. 15).27-31,67-70 In the aggregate, these deficits are extreme examples of the post-intensive care syndrome, and they represent an important aspect of the impact of ARDS.
PATHOLOGY AND PATHOPHYSIOLOGY
It can be useful to distinguish between the early phases of lung injury and subsequent events, as the early phases are characterized by exuberant inflammation and the later phases by repair and fibrotic mechanisms.38,71,72 By light microscopy, the early appearance is of interstitial and alveolar edema, capillary congestion, and intra-alveolar hemorrhage with minimal evidence of cellular injury (Fig. 52-3). By electron microscopy, changes of endothelial cell swelling, widening of intercellular junctions, increased numbers of pinocytotic vesicles, and disruption and denudation of the basement membrane are prominent. Inflammatory cell infiltration of the lung interstitium may be seen (particularly in ARDS complicating sepsis or trauma) as well as neutrophil sequestration in alveolar capillaries. During this early exudative phase of diffuse alveolar damage (DAD), pulmonary edema and its clinical effects are most pronounced (see Fig. 52-3). It is also a time when manipulations to decrease the rate of edemagenesis are most likely to have an impact as discussed below.
FIGURE 52-3
A schematic representation showing the time course of evolution of the acute respiratory distress syndrome (ARDS). During the early or exudative phase, the lesion is characterized by a pulmonary capillary leak with interstitial and alveolar edema and hemorrhage, followed by hyaline membrane formation. Within as short a period of time as 7 to 10 days, a proliferative phase may appear with marked interstitial and alveolar inflammation and cellular proliferation, which is followed by fibrosis and disordered healing (see text for discussion). (Reproduced with permission from Katzenstein AA, Askin FB. Surgical Pathology of Non-Neoplastic Lung Diseases. 2nd ed. Philadelphia, PA: Saunders; 1990.)

Over the ensuing days, hyaline membrane formation in the alveolar spaces becomes prominent. Hyaline membranes contain condensed fibrin and plasma proteins. Intra-alveolar activation of the coagulation system results in the formation of the fibrin, while plasma proteins are deposited in the alveolar space as part of the inflammatory exudate that leaks across the alveolar-capillary membrane. Inflammatory cells become more numerous within the lung interstitium. As the process of DAD progresses, there is extensive necrosis of type I alveolar epithelial cells. If the patient with ARDS does not recover or die during the first week, they may have a prolonged course of illness, termed late-phase ARDS.
The later phase of ARDS is dominated by disordered healing. This can occur as early as 7 to 10 days after initial injury and may eventually result in extensive pulmonary fibrosis. This has been termed the proliferative or fibroproliferative phase. Type II alveolar cells proliferate along alveolar septae and the alveolar walls; fibroblasts and myofibroblasts become more numerous. Evidence of lung flooding is less prominent and may be minimal at this point. Changes in the clinical manifestations of ALI and ARDS parallel the changes in pathology. One study found that patients in late-phase ARDS typically had a large dead space fraction, a high minute ventilation requirement, progressive pulmonary hypertension, slightly improved intrapulmonary shunt that is less responsive to PEEP, and a further reduction in lung compliance.73
PATHOGENESIS
A number of closely interrelated pathophysiologic mechanisms and systems contribute to the development of ARDS (Fig. 52-4). Dysregulated inflammation, excess oxygen radicals, activation of coagulation and impaired fibrinolysis, platelet and immune cell activation, and loss of alveolar barrier integrity are central mechanisms in the pathogenesis of ARDS.34,38,39,74-76 In addition to ongoing inflammation and oxidation, factors specific to apoptosis, edema fluid resolution, fibrosis, and repair are likely to be important in the resolving and late phases of ARDS.34,38,39,74,76
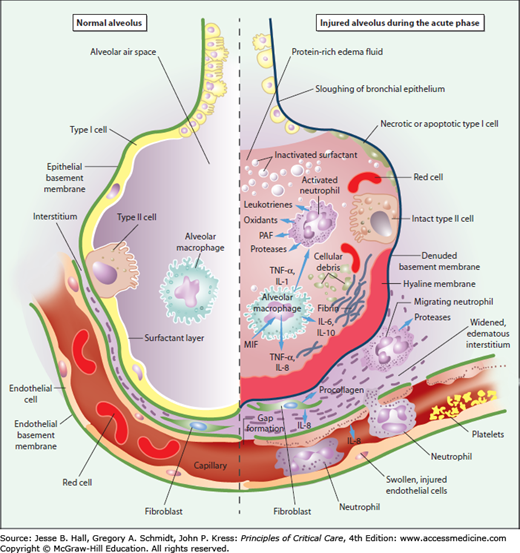
FIGURE 52-4
Schematic representation of the alveolus under normal conditions (left-hand side) and during development of acute lung injury (ALI) and the acute respiratory distress syndrome (ARDS) (right-hand side). In the acute phase of the syndrome (right-hand side), there is sloughing of both the bronchial and alveolar epithelial cells, with the formation of protein-rich hyaline membranes on the denuded basement membrane. Neutrophils are shown adhering to the injured capillary endothelium and marginating through the interstitium into the air space, which is filled with protein-rich edema fluid. In the air space, an alveolar macrophage is secreting cytokines such as interleukins-1, -6, -8, and -10 (IL-1, 6, 8, and 10), and tumor necrosis factor-α (TNF-α), which act locally to stimulate chemotaxis and activate neutrophils. Macrophages also secrete other cytokines, including interleukins-1, -6, and -10. Interleukin-1 can also stimulate the production of extracellular matrix by fibroblasts. Neutrophils can release oxidants, proteases, leukotrienes, and other proinflammatory molecules, such as platelet-activating factor (PAF). A number of anti-inflammatory mediators are also present in the alveolar milieu, including interleukin-1–receptor antagonist, soluble tumor necrosis factor receptor, autoantibodies against interleukin-8, and cytokines such as interleukins-10 and -11 (not shown). The influx of protein-rich edema fluid into the alveolus has led to the inactivation of surfactant. MIF denotes macrophage inhibitory factor. (Reproduced with permission from Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. May 4, 2000;342(18):1334-1349.)
ARDS develops after both systemic inflammatory insults and directly as a result of pulmonary infections and injury. Inflammatory mechanisms, when properly regulated, are important to containment and healing from insults such as pneumonia, aspiration, and systemic infection and injury. However, for reasons that are incompletely understood, in ARDS these same mechanisms become dysregulated, leading to pulmonary accumulation of immune cells and platelets, unopposed oxidant injury to lung tissues, enhanced coagulation and impaired fibrinolysis, and disruption of the normal function of the alveolar-capillary membrane.
This early response to tissue damage or pathogens leads to early release of inflammatory cytokines and chemokines by resident lung cells, including dendritic cells. Two of the major early proinflammatory cytokines are tumor necrosis factor-α (TNF-α) and interleukin-1 (IL-1), both of whose production can be increased by hypoxia.77-79 These cytokines have similar effects in initiating and propagating inflammation.77,80-83 Their actions include (1) recruitment, differentiation, amplification, and localization of macrophages to the lung parenchyma; (2) stimulation of other inflammatory cytokines such as interleukins-6 and -8 (IL-6 and IL-8); and (3) adherence of neutrophils to endothelium (see Fig. 52-4).77,84-86 Both BAL and plasma fluids consistently have been reported to contain elevated levels of TNF-α and IL-1 in ARDS patients.87-91 Macrophages that have been stimulated by TNF-α and IL-1, in turn produce IL-6, which has diverse functions. Like TNF-α and IL-1, persistently elevated IL-6 levels have been associated with an increased risk of death in ARDS patients.92 In addition, in the ARDSNet ARMA trial the lung-protective treatment arm that had decreased mortality showed a greater attenuation in inflammatory cytokines (IL-6) and chemokines (IL-8) compared to the higher-tidal-volume arm.3,93
Recently, it has been increasingly recognized that innate immune mechanisms in response to pathogens or tissue damage are key to this early inflammatory response. Innate immune pattern recognition receptors that are either localized in the cell membrane (eg, Toll-like receptors, TLRs) or in the cytoplasm (eg, Nod-like receptors, NLRs) can become activated in the early stages in ARDS. Both danger- and pathogen-associated molecular pattern (DAMPs and PAMPs) recognition play an important role.94,95 Stimuli for activation of these pathways include bacterial and viral pathogens,96 lysosomal disruption,97 neutrophil- or mitochondria-derived reactive oxygen species (ROS),98,99 and cell apoptosis.99,100 Activation of these pathways can lead to formation of inflammasomes, which are proinflammatory macromolecular complexes that activate caspase-1, resulting in IL-1β and IL-18 release.101 Inflammasome activation has been demonstrated in clinical studies, solidifying the key role in ARDS pathogenesis. Furthermore, recent studies suggest that an imbalance between IL-1β and the IL-1 receptor antagonist (IL1-RA) is important in ARDS risk and outcome.102 Genetic regulation of this pathway is likewise implicated in ARDS pathophysiology.53
Stimulated dendritic cells and tissue macrophages in the lung also produce IL-23, which in turn induces production of interleukin-17 (IL-17) by T-helper cells. IL-17A is the original member of what is now the IL-17 family of cytokines.103 IL-17 is the major product of TH17 cells, which are a helper T-cell subtype characterized by enhanced inflammatory response, particularly in mucosal immunity and lung injury.104-106 IL-17A is thought to have pleiotropic effects; however, promotion of neutrophil chemotaxis is dominant.107 Neutrophil migration into the airspaces is evident in the early histopathology of ARDS and is further mediated by IL-8 and intercellular adhesion molecules (eg, intercellular adhesion molecule-1 [ICAM]).108,109 BAL fluid of subjects with ARDS reveals a predominance of neutrophils.110 Conversely, recovery is associated with resolution of neutrophilia.111 Normally, neutrophils become apoptotic and are then removed by macrophages. Impaired neutrophil apoptosis may be an important mechanism in ARDS,112 and elevated levels of granulocyte-macrophage colony-stimulating factor (GM-CSF) may play a role in this mechanism.113-115
Neutrophils act on pathogens through intracellular and extracellular mechanisms, including phagocytosis, degranulation of toxic proteins contained in neutrophils granules, and the formation of neutrophil extracellular traps (NETs).116 NETs are composed of a core DNA element that affixes histones and contents of neutrophils granules. Neutrophil degranulation and NET formation are intended to immobilize and destroy pathogens; however, these mechanisms can lead to substantial tissue damage in ARDS. Recently, NETs have been shown to be key mediators of transfusion associated ALI.117
Neutrophils in the alveolar and interstitial spaces may result in lung injury due to neutrophil-mediated release of reactive oxygen species (ROS) and resulting oxidant stress. Oxidant stress refers to an imbalance between endogenously produced oxidants and endogenous antioxidants. Products of oxidant stress include superoxide, hydroxyl radicals, and peroxynitrite.118 Although some levels of ROS may be important in normal homeostasis, ROS are highly unstable and react quickly with surrounding proteins, DNA, and lipids, resulting in molecular damage.118
A widely held hypothesis is that excessive oxidant stress contributes to the pathogenesis of ALI.119-126 ROS can be generated by neutrophils (polymorphonuclear [PMN] cells),119,121,127 or by the pulmonary endothelium.128,129 Both PMN-mediated and endothelial generation of ROS may be important to initiation and development of lung injury. Evidence from clinical studies supporting excess ROS in ALI and ARDS include findings of increased hydrogen peroxide in exhaled breath of ARDS patients130,131; decreased levels of glutathione in lung lavage fluids of ARDS patients132,133; increased nitrotyrosine and chlorotyrosine in lavage fluids of ARDS patients134; increased lipid peroxidation products in plasma135,136; increased protein carbonyl levels137; increased plasma hypoxanthine138; increased levels of nitrated fibrinogen in the plasma of ALI/ARDS patients139; and increased serum ferritin levels in ARDS patients compared with at-risk patients.140,141 Although some of these findings may be nonspecific for ARDS,74 taken together, they make a strong case for the role of oxidant stress in the pathogenesis of ARDS. In addition, evidence of higher levels of oxidant stress in nonsurvivors of ARDS is illustrated by lower levels of plasma thiol groups,142 and higher levels of hydrogen peroxide in urine of ARDS patients who are nonsurvivors.143
The histopathology of the early exudative phase of ARDS is notable for diffuse alveolar fibrin deposition in the form of hyaline membranes (see Fig. 52-3).71,72,109,144,145 In addition, a shift in the procoagulant and anticoagulant balance to favor coagulation has been demonstrated in the BAL fluid of patients with ARDS.146-150 Furthermore, there are likely important links between the coagulation system activation and activation of the inflammatory system. For example, TNF-α and IL-1 can act synergistically to produce a procoagulant state through effects on tissue factor, thrombomodulin, and plasminogen activator inhibitor151-153 and there exists an independent relationship between markers of inflammation, neutrophil migration, and coagulation and fibrinolysis and mortality in ALI patients.154 Although prior studies found plasma protein C levels are decreased in ALI155 and decreased levels are associated with worse outcomes,156 a small randomized trial of activated protein C for use in patients with ALI did not appear to improve outcomes.157 Furthermore, in 2011, activated protein C was withdrawn from the market for patients with severe sepsis after a large trial failed to demonstrate efficacy.158 Despite strong scientific rationale, interventions aimed at correcting abnormalities in coagulation and fibrinolysis in critically ill patients have not improved outcomes to date, although further research is warranted.
Dysregulated inflammation and direct injury can lead to disruption of the alveolar-capillary membrane. Several key mediators of endothelial permeability, inflammation, and angiogenesis have been suggested in ARDS pathogenesis. For example, angiopoietin-2 (Ang-2) and vascular endothelial growth factor (VEGF) are mediators of endothelial permeability and angiogenesis. Ang-2 expression inhibits angiopoietin-1, an endothelial stabilizing factor. Increased levels of Ang-2 have been associated with the development of ALI and adverse outcomes in those who develop ALI159 and variation in the ANGPT2 alters risk of development of ALI.160 As a marker of endothelial injury, von Willebrand factor antigen (vWF) predicted the development of ALI in several at-risk populations,161,162 although these findings have not been replicated consistently. Likewise, a recent study suggests that lower levels of endocan, a proteoglycan expressed by endothelial cells, may be associated with development of trauma-associated ALI as the protective effect of endocan to inhibit leukocyte recruitment via ICAM-1 may be reduced.163
Furthermore, recent research into alveolar barrier integrity suggests several additional mechanisms with potential therapeutic targets. Recent evidence supports a role of the balance between angiotensin-converting enzymes 1 and 2, in modulating inflammatory lung injury in pulmonary and systemic infections.164,165 Vascular endothelial cadherin (VE-cadherin) is a junction protein that is involved in maintenance of barrier integrity in lung microvessels.166 Instability in complexes of VE-cadherin and catenin may affect capillary leak and inflammatory cell extravasation.167,168 The recently recognized Robo4/Slit signaling system is expressed on endothelial cells and serves to stabilize the endothelial barrier in the lung. A member of this system, Slit2N reduces permeability in microvascular endothelial cells caused by inflammatory cytokines, influenza, and sepsis.169 Sphingosine-1-phosphate (S1P) is a lipid present in high concentrations in plasma that binds to receptors on endothelial cells to enhance pulmonary endothelial barrier integrity in a variety of pathogenic conditions.170,171 Future research into the regulation of different S1P receptors during the time course of lung injury is necessary.172
FIBROSIS, RESOLUTION, AND REPAIR MECHANISMS
Although there is considerable temporal overlap of the exudative and inflammatory phases (see Fig. 52-3), the later phases of ARDS are characterized by accumulation of matrix and cells in the interstitial and alveolar spaces, contributing to disruption of the alveolar architecture.71,72 Recovery from ARDS requires resolution of this phase of the tissue injury–repair spectrum to restore the normal lung architecture. Clinical studies of ARDS patients indicate that the alveolus is repaired under concurrent and ongoing inflammation.78 Soluble collagen precursor, type III procollagen peptide, in the edema fluid of early ARDS can differentiate between subjects who will have longer or shorter courses of ARDS and fibroblast mitogenic activity in BAL fluid is lower in survivors of ARDS than nonsurvivors,173 suggesting that fibroproliferation begins early in the course of lung injury.174,175
An orchestrated balance of different growth factors, cytokines, and chemokines occurs in the fibroproliferative and reparative phases of ALI.74,176 A variety of animal, basic, and clinical studies have demonstrated the importance of transforming growth factor-β (TGF-β) to fibroproliferation in the lung.177,178 A key step in fibroproliferation involves the activation of TGF-β from its latent form. This can be accomplished through interaction with a variety of mediators, including matrix metalloproteinases (MMPs), thrombospondin, plasmin, acid environments, α2-macroglobulin, and ROS.177 In addition, TGF-β has extensive interactions with inflammatory cytokines, chemokines, and interferon-γ in regulating fibrosis. Leptin, a protein secreted by adipose tissue that decreases hunger through a signal to the brain, is also an inflammatory mediator that has been implicated in fibrogenesis. Recently, leptin resistance was identified as a potential protective factor for the development of ALI and an explanation for the observed association between diabetes and decreased risk for ALI resistance.179
Other important modulators of the remodeling of fibrosis include matrix and cell surface proteoglycans and glycosaminoglycans180-182; matricellular proteins that affect cell adhesion183,184; matrix metalloproteases185; and the balance of coagulation and fibrinolysis (particularly through the actions of plasminogen activator inhibitor-1 [PAI-1]).137 The interplay of these mediators with the potential for targeted therapies is the focus of current investigations.
In addition to remodeling of fibrosis, resolution of lung injury in the ARDS patient requires several overlapping reparative mechanisms, including deactivation and clearance of inflammatory cells from the alveolar space, repair of the epithelial barrier, and resorption of edema fluid. For the most part, these mechanisms are occurring during overlapping time intervals as the injured lung attempts to repair and restore its physiological state.
Resolution of inflammation requires local production of counterregulatory inhibitory molecules of the inflammatory response (including TLR, NOD, cytokine inhibitors). Molecules important in the resolution phase are produced locally and include lipoxins, resolvins, and D-series prostaglandins, among others.186 An important part of this process involves clearance of apoptotic neutrophils and other inflammatory debris from the alveolar space my alveolar macrophages.187 Regulatory T cells play a key role in this process, forming an “immune synapse” with alveolar macrophages to help orchestrate the process.188
Recovery of the alveolar epithelium requires repopulation of the alveolus with type I epithelial cells. At baseline, the human lung has little cell turn over compared to some organ systems; however, the lung can rapidly regenerate new cells after injury.189 Epithelial cells may arise from differentiation of Type II pneumocytes, or from recently described human lung stem cells.189,190 Recovery of epithelial cells may require epithelial-mesenchymal interaction with triggering of developmental pathways, such as Wnt signaling pathways.189
Additionally, epithelial protection and recovery have recently been shown to rely on interactions with innate lymphoid cell (ILC) populations, which are similar to CD4+ T-helper cells and play critical roles in antipathogen immunity, regulation of inflammation, and promotion of wound healing and tissue repair at barrier surfaces.190-199 Recently, ILC2 cells have been shown to have a major role in maintaining alveolar homeostasis during injury response in the lung.200 In the presence of pathogens, IL-33 and IL-25 induce lineage negative ILC2 cells to produce amphiregulin, which binds to the EGFR receptor to protect epithelium from damage by maintaining barrier function, enhancing epithelial growth and repair, and maintaining oxygenation.200
Considerable evidence supports the role of active transport of sodium and water by the pulmonary epithelium as a means to remove pulmonary edema from alveoli.201 This process appears to be regulated by epithelial sodium channels predominantly on alveolar type I cells, potentially stimulated by catecholamines.202,203 Furthermore, the directional flow of fluid across the alveolus may be altered in conditions of hypoxia,204,205 as well as by reactive nitrogen and oxygen species.206
There is considerable evidence of epithelial injury and dysfunction in clinical ARDS. Biomarkers of epithelial injury have demonstrated association with clinical ARDS risk and/or outcome including surfactant proteins,207,208 Clara cell protein secreted by the epithelial Clara cell,209 and the receptor for advanced glycation end products (RAGE).210 Clinical studies in ventilated patients have illustrated impaired fluid transport in ARDS when compared with hydrostatic pulmonary edema.38,211,212 In addition, subjects who upregulate alveolar fluid clearance have a lower hospital mortality.212,213 However, a recent trial designed to augment the rate of alveolar edema clearance through the use of β-adrenergic receptor agonists was terminated early due to futility, indicating that restoration of the epithelial cell barrier is essential to fluid resorption.214
APPROACH TO DIAGNOSIS OF ALI AND ARDS
AHRF has many etiologies besides ALI and ARDS (Table 52-3). However, the bedside appearance of patients with various forms of AHRF is remarkably similar. Marked tachypnea and dyspnea are invariably present. Physical examination reveals diffuse crackles in cases of cardiogenic pulmonary edema and focal findings of consolidation in cases of lobar pneumonia. Cardiogenic pulmonary edema may be accompanied by evidence of airflow obstruction, including wheezing and hypercapnia.215 The presence of crackles, a radiologic appearance of high-pressure edema (see below), and hypoxemia refractory to oxygen therapy, all suggest cardiogenic pulmonary edema as the primary process. Cough and purulent sputum are hallmarks of infectious processes, while copious clear or pink-colored airway secretions result from fulminant (“flash”) cardiogenic pulmonary edema.
Differential Diagnosis of Acute Hypoxemic Respiratory Failure (AHRF)
|
Distressed patients with AHRF typically have initial room air arterial blood gas results with PaO2 in the 30 to 55 mm Hg range and pulse oximetry less than 85% of arterial O2 saturation. If supplemental oxygen by mask or cannula raises arterial saturation to above 95%, a large intrapulmonary shunt is unlikely. Other causes of respiratory distress should then be considered, including airways disease, pulmonary embolus, or severe metabolic acidosis. Failure to achieve >95% saturation of arterial blood with supplemental oxygen indicates the presence of a large right-to-left shunt. The specific process should be investigated via physical examination and chest radiograph. In the rare instances that the chest radiograph is entirely clear of alveolar infiltrates, one should consider that the blood gas data are erroneous, that there is an anatomic right-to-left shunt at another site (eg, pulmonary arteriovenous malformations or intracardiac shunt), or that there is continued perfusion of an unventilated lung due to recent complete or nearly complete occlusion of its main bronchus (but before the lung has collapsed due to absorption atelectasis) (see Table 52-3).
The differential diagnosis of ALI and ARDS (ie, AHRF with diffuse pulmonary infiltrates consistent with pulmonary edema in the absence of a cardiac etiology) includes a variety of disorders and etiologies. Identifying the etiologies of the diffuse infiltrates is important because specific treatments exist for several of these conditions (eg, acute eosinophilic pneumonia or diffuse alveolar hemorrhage). Table 52-4 lists the major clinical and diagnostic characteristics of these disorders.
Differential Diagnosis of Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome (ARDS)
| Disorder | Characteristics | Comment |
|---|---|---|
| Pulmonary edema due to left heart failure | History of cardiac disease, enlarged heart on chest radiograph, third heart sound (S3) | Rapid improvement with diuresis and/or afterload reduction |
| Noncardiogenic pulmonary edema | History of one or more precipitating causes (see Table 38-2), crackles absent or not prominent, normal cardiac size on chest radiograph | Usual etiology for ALI and ARDS: Rarely some patients with ALI or ARDS have no obvious precipitating cause |
| Diffuse alveolar hemorrhage (DAH) | Often associated with autoimmune diseases (eg, vasculitis) or following bone marrow transplantation; often patients do not have bloody sputum; renal disease or other evidence of systemic vasculitis may be present; hemosiderin-laden macrophages in bronchoalveolar lavage (BAL) fluid can confirm diagnosis of DAH; may respond to apheresis, corticosteroids, or cyclophosphamide, depending on etiology | May meet diagnostic criteria for ARDS, but has different pathophysiology and management |
| Acute eosinophilic pneumonia | Cough, fever, pleuritic chest pain, and myalgia are often present; patients often do not have peripheral blood eosinophilia, but generally have >15% eosinophils in BAL fluid; usually responds rapidly to high-dose corticosteroid therapy | May meet diagnostic criteria for ARDS, but has different pathophysiology and management |
| Lupus pneumonitis | Usually associated with active lupus; may respond to high-dose corticosteroid therapy or cyclophosphamide | May meet diagnostic criteria for ARDS, but has different pathophysiology and management |
| Acute interstitial pneumonia (AIP) | Slower onset than ARDS (over 4-6 weeks) with progressive course; however, it may present in an advanced state, mimicking ARDS | Associated with >90% mortality; AIP includes Hamman-Rich syndrome |
| Pulmonary alveolar proteinosis (PAP) | Slower onset than ARDS (over 2-12 months) with progressive course; can be treated with whole lung lavage | Characteristic “crazy paving” pattern on high-resolution computed tomography scan |
| Bronchiolitis obliterans organizing pneumonia (BOOP) or cryptogenic organizing pneumonia | May be precipitated by viral syndrome; slower onset than ARDS (over >2 weeks) with progressive course; however, it may present in an advanced state, mimicking ARDS; may respond to high-dose corticosteroid therapy | |
| Hypersensitivity pneumonitis | Typically slower onset than ARDS (over weeks) with progressive course; however, it may present in an advanced state, mimicking ARDS; may respond to high-dose corticosteroid therapy and removal from offending agent | |
| Leukemic infiltration | May be rapid in onset during active disease states; usually leukemia is clinically apparent | |
| Drug-induced pulmonary edema and pneumonitis | May follow use of heroin, other opioids, overdose of aspirin, tricyclic antidepressants, or exposure to paraquat | May progress to overt ARDS |
| Acute major pulmonary embolus (PE) | Occurs acutely, occasionally accompanied by severe hypoxemia that may be resistant to O2 therapy like ARDS, and by hypotension, requiring pressors, mimicking ARDS with sepsis; patients typically have risk factors for acute PE and may not have common precipitating causes of ARDS | Chest radiograph in ARDS should have bilateral infiltrates consistent with pulmonary edema; chest radiograph in acute major PE may have unilateral or no infiltrates; acute major PE needs a confirmatory study (eg, pulmonary angiogram) |
| Sarcoidosis | The onset is not acute, but its clinical recognition may be; oxygenation is often impaired and the chest radiograph can be diffusely abnormal | Historical features and the frequent presence of hilar adenopathy in sarcoidosis usually eliminate confusion with ARDS |
| Interstitial pulmonary fibrosis | The onset is not acute, but its clinical recognition may be; oxygenation is often impaired and the chest radiograph can be diffusely abnormal | Prior chest radiographs and a history of chronic and progressive dyspnea characterize the collection of diseases causing interstitial pulmonary fibrosis |
Clinical Setting: The clinical setting in which the disorder develops can provide important diagnostic information. Cardiogenic edema is most often accompanied by systolic left ventricular or valvular dysfunction, and the abnormal heart sounds and murmurs associated with each should be sought. Electrocardiographic (ECG) and serum enzyme evidence of ischemia should be considered and suggest an obvious cause for cardiogenic edema. Review of intravascular volume administration often will supply information suggesting the explanation for pulmonary edema in patients with left ventricular or renal dysfunction.
ALI and ARDS commonly arise in a typical clinical context (see Table 52-2). Sepsis, pneumonia, trauma, transfusion of blood products, and acid aspiration account for the majority of cases of ALI and ARDS.3,4,13,38 Less common causes include pancreatitis, near-drowning, leukoagglutination reactions, lung infections with viral agents or Pneumocystis jiroveci, fat embolism syndrome, and drug toxicities.38
Chest Radiograph: The chest radiograph is a simple and widely available test used to assess patients with AHRF. Unfortunately, the accuracy of the routine radiograph in distinguishing hydrostatic from increased permeability edema is not high.216,217 Criteria that have been suggested to support a diagnosis of hydrostatic edema include increased heart size, increased width of the vascular pedicle, vascular redistribution toward upper lobes, septal lines, and a centrifugal pattern of spread with a perihilar bat’s-wing distribution of the edema. The lack of these findings and patchy peripheral infiltrates that extend to the lateral lung margins suggest ARDS. However, all these signs overlap, and in the best of hands this test is unlikely to yield better than a 60% to 80% accuracy of diagnosis when applied without other diagnostic tools.216
Echocardiography: Echocardiography is a useful noninvasive diagnostic tool to obtain information regarding cardiovascular function218,219 and may provide useful diagnostic and/or therapeutic information in ALI patients.220 Left ventricular dilation, regional or global wall motion abnormalities, and substantial mitral regurgitation on Doppler imaging support a diagnosis of cardiogenic edema. A heart with echocardiographically normal dimensions and function (both systolic and diastolic) in a patient with pulmonary edema suggests pulmonary vascular leakage, although prior ventricular or valvular dysfunction with intercurrent resolution of the high pulmonary vascular pressures predisposing to cardiogenic edema must be kept in mind.
In patients with ALI, pulmonary vascular dysfunction, as measured by transpulmonary gradient or pulmonary vascular resistance index derived from pulmonary artery catheter data, is common and predictive of mortality.221 However, isolated echocardiography-derived measures of pulmonary vascular dysfunction (eg, pulmonary artery systolic pressure, cardiac index) have not been found to be predictive of mortality,221,222 Tricuspid annular plane systolic excursion, a echocardiography-derived measure of right ventricular ejection fraction, may prove to be a useful prognostic tool in ALI patients. Using contrast-enhanced echocardiography, a prospective study found that moderate to large shunting occurs across a patent foramen ovale in approximately 20% of ARDS patients and oxygenation was less responsive to increased PEEP in patients with evidence of intracardiac shunting.220
Pulmonary Artery Catheterization: Between its development in 1970 and 2006, when the NIH NHLBI ARDSNet clinical trial was published that found that pulmonary artery catheterization (PAC)–guided therapy was associated with increased complications without clinical benefit,22 PAC was frequently performed in patients with pulmonary edema.223-225 Even before the results of the trial were published, based on prior studies suggesting that the technology itself and/or misinterpretation of the information provided led to worse patient outcomes,226,227 PAC use had substantially decreased.228
At present, PAC-guided therapy cannot be recommended as a routine procedure in patients with ALI/ARDS given increased complications and increased cost without a proven benefit.22,229 Furthermore, with increased use, availability, and experience with echocardiography and alternative means to assess for fluid responsiveness (eg, passive straight leg raise test), and less experience with interpretation of the PAC, it is recommended that clinicians use noninvasive methods to address specific questions regarding ventricular function, the adequacy of volume resuscitation, and the adequacy of cardiac output and oxygen saturation of mixed venous blood.
Furthermore, as noted previously, the specific Ppw that the AECC definition (see Table 52-1) used as the criterion to distinguish noncardiogenic from cardiogenic pulmonary edema was an arbitrary decision based on physiologic experiments, tradition, and volume resuscitation practices circa 1992, and nearly one in three patients with ALI will have a Ppw that exceeds the 18 mm Hg threshold.22 In the mechanically ventilated patient with normal lung function and serum oncotic pressure, cardiogenic edema is typically associated with a Ppw of 28 mm Hg or above.230 However, lower plasma oncotic pressure (eg, due to hypoalbuminemia) will result in pulmonary edema at lower intravascular pressure values.231
Acute eosinophilic pneumonia is a rare disorder that is characterized by diffuse AHRF due to eosinophilic infiltrates in the lungs.232,233 It is notable for its responsiveness to corticosteroid therapy. When the precipitating cause for ARDS is unclear, it is recommended to perform a bronchoalveolar lavage and measure the percentage of eosinophils in the lavage fluid.232 Lavages can generally be done safely in many patients with ALI and ARDS except those with the lowest values of PaO2 : FiO2 or hemodynamic instability.234,235
Likewise, a bedside bronchoscopy with BAL can be diagnostic for diffuse alveolar hemorrhage (DAH) or identifying a causative microbiologic organism. In the former case, the bronchoscopy may or may not reveal fresh blood in the trachea and major bronchi. However, BAL generally produces a bloody return, which may deepen in red color as the lavage continues. DAH occurs commonly in the first week or two post-bone marrow transplantation.236,237 DAH also occurs in association with a variety of vasculitic disorders. These include Goodpasture syndrome, Wegener granulomatosis, systemic lupus erythematosus, and antiphospholipid antibody syndrome (see Chap. 126).238-243 Finally, DAH may also result from inhalation of crack cocaine.238 For this cause of DAH, careful history taking and sending the patient’s urine for toxicology analysis for cocaine may help determine the etiology.
APPROACH TO TREATMENT OF PATIENTS WITH ALI AND ARDS
A key early step in treating patients with ALI and ARDS is to identify and treat the precipitating cause or causes of the ALI and ARDS as well as any other serious and life-threatening comorbidities (Fig. 52-5). The ventilatory and other supportive management of ALI and ARDS is inadequate if not accompanied by aggressive attempts at diagnosis and treatment of the precipitating cause(s) (Table 52-2). Because ARDS is a syndrome based on nonspecific radiographic and physiologic criteria (Table 52-1), making the diagnosis of ALI or ARDS is not equivalent to diagnosing the patient’s underlying problem. Not appreciating this seemingly obvious fact will delay diagnostic procedures in these patients and may delay therapy of a potentially treatable underlying disorder (Table 52-5).
Treatable Precipitating Causes of Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome (ARDS)
| Infectious etiologies |
| Bacterial or other sepsis responsive to antimicrobial therapy |
| Diffuse bacterial pneumonias (eg, Legionella species) |
| Diffuse viral pneumonias (eg, cytomegalovirus, influenza A) |
| Diffuse fungal pneumonias (eg, Candida and Cryptococcus species) |
| Pneumocystis jiroveci (carinii) pneumonia |
| Other diffuse lung infections (eg, miliary tuberculosis) |
| Noninfectious etiologies |
| Diffuse alveolar hemorrhage post-bone marrow transplant |
| Diffuse alveolar hemorrhage due to vasculitis (eg, Goodpasture syndrome) |
| Acute eosinophilic pneumonia |
| Lupus pneumonitis |
| Toxic drug reactions (eg, aspirin, nitrofurantoin) |
For example, although appropriate supportive therapy may transiently stabilize a patient with ARDS due to sepsis from an abdominal abscess, if clinicians delay performing diagnostic tests such as abdominal CT scan or ultrasonography of the biliary tract in a timely manner, the underlying source of sepsis will go undiagnosed and the patient will eventually deteriorate. Likewise, the timely start of empiric antimicrobial therapy in patients with ALI or ARDS associated with severe sepsis or septic shock is as important as a timely diagnostic workup (see Chaps. 61 and 62). Finally, if the precipitating cause of ALI and ARDS is unclear, one should consider performing early fiberoptic bronchoscopy to obtain bronchoalveolar lavage for cytologic and microbiologic analyses, or in selected cases, surgical lung biopsy.
Maintaining Adequate Arterial Oxygenation: The hallmark respiratory abnormality of ALI and ARDS is hypoxemia that is resistant to oxygen therapy. This is due to the presence of a large right-to-left intrapulmonary shunt arising from fluid-filled and collapsed alveoli (see Fig. 52-1). Maintaining adequate arterial oxygenation is a goal given high priority by both traditional and more recent approaches to ventilator management, as noted below. One should use sufficient PEEP to reduce the right- to-left shunt to oxygenate the patient. By this approach, clinicians should be able to avoid prolonged exposure of such patients to potentially toxic concentrations of high inspired oxygen (eg, FiO2 of 0.7 and above). PEEP improves arterial oxygenation, primarily by recruiting collapsed and partially fluid-filled alveoli and thereby increasing the functional residual capacity (FRC) at end expiration.244,245 PEEP redistributes alveolar fluid into the interstitium,246 which should also improve oxygenation.
Noninvasive Ventilation: Assisted ventilation is generally provided via an endotracheal tube, but in selected cases noninvasive ventilation (NIV) may be successful247,248 (see Chap. 44). Although NIV seems to be useful in respiratory failure in immunocompromised hosts,247 the failure rate approaches 50% in such instances.249 As such, it is generally not a good choice for most patients with ALI and ARDS and caution is advised if it is attempted. This is because ARDS typically has a long course and is often associated with hemodynamic instability, coma, and multiorgan system failure (including ileus).250
Comparison of Traditional and Current Approaches to Ventilator Management
Goals and Priorities of the Traditional Approach The traditional approach to ventilator management of patients with ALI and ARDS gave high priority to these goals: (1) to maintain arterial O2 saturation (O2 saturation) above 88% to 90% to provide for adequate tissue oxygenation while trying to minimize lung injury due to high concentrations of inspired oxygen (oxygen toxicity); (2) to provide sufficient ventilation to keep arterial pH and PaO2 within normal limits251 (Fig. 52-6).
FIGURE 52-6
Schematic illustration that demonstrates how traditional and lung-protective approaches to mechanical ventilation of patients with ARDS have different priorities. The traditional approach gives higher priority to keeping arterial pH and  normal (and possibly to keeping the patient more comfortable) than the lung-protective approach, which gives higher priority to prevention of ventilator-induced lung injury (VILI). Plateau pressure = static end-inspiratory pressure in the alveoli.
normal (and possibly to keeping the patient more comfortable) than the lung-protective approach, which gives higher priority to prevention of ventilator-induced lung injury (VILI). Plateau pressure = static end-inspiratory pressure in the alveoli.
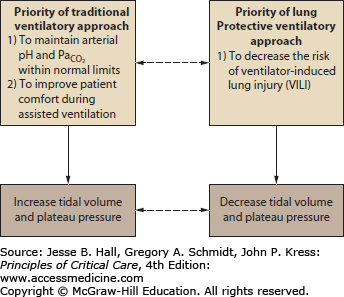
To achieve the first goal, clinicians applied various levels of PEEP. This use of PEEP was first described in the initial description of ARDS by Ashbaugh and coworkers in 1967.10 Clinicians increased levels of PEEP in order to decrease Fi
Related posts:






Full access? Get Clinical Tree


