KEY POINTS
Prerenal azotemia and acute tubular necrosis account for the overwhelming majority of hospital-acquired acute kidney injury cases, whereas acute glomerulonephritis and vasculitides are relatively more common causes of acute kidney injury developing outside the hospital.
Acute kidney injury occurs in at least 10% to 30% of patients admitted to an ICU, and severe AKI is associated with a mortality rate of about 50%, despite advances in supportive care and technology.
Traditionally, the most important diagnostic classification to be made in the evaluation of patients with acute kidney injury is based on the site of the renal lesion (pre-, intra-, or postrenal).
Since there are few specific therapies available in patients with established acute tubular necrosis, the major clinical focus is on prevention of AKI by identification of subjects at highest risk.
All aspects of treatment of acute tubular necrosis, including renal replacement therapy, are basically supportive. The nondialytic measures of greatest importance are maintenance of nutritional, volume, and electrolyte homeostasis.
Acute renal failure (ARF) is defined as a rapid decline (over hours to days) in glomerular filtration rate (GFR). This manifests as a rapid increase in blood urea nitrogen (BUN; “azotemia”) and serum creatinine, and may or may not be accompanied by a decline in urine output.1 The concept of acute renal failure has undergone significant change over the last number of years. Lack of standardization in the definition of acute renal failure and the emergence of evidence that even small increases in serum creatinine are associated with increased mortality has led to widespread adoption of diagnostic criteria for the term acute kidney injury (AKI).2 AKI has largely replaced the term acute renal failure (ARF). It is a syndrome that includes minor degrees of injury as well as more severe renal failure, and does not allude to the mechanism of injury. Glomerular filtration rate (GFR) is the best measure of kidney function, but is not easily measured in clinical practice. A change in serum creatinine or urine output is used as a marker for a change in GFR and forms the basis for the various diagnostic criteria for AKI.
A number of classification systems for AKI exist; the most widely validated is the RIFLE system.3,4 This classification system was proposed by the Acute Dialysis Quality Initiative (ADQI) in 2004.5 The acronym RIFLE represents three severity of injury classes: risk, injury, and failure, and two outcomes: loss of function and end-stage renal disease (Fig. 97-1). The severity of injury is defined by the magnitude of increase in serum creatinine from a baseline value (within a 7-day period or less), or a reduction in urine output for a defined period of time. The outcomes are defined by the duration of kidney injury. Criticisms of the RIFLE classification include the need for a baseline creatinine value to define a case of AKI, and a lack of clarity concerning the effect of RRT requirement on AKI staging.
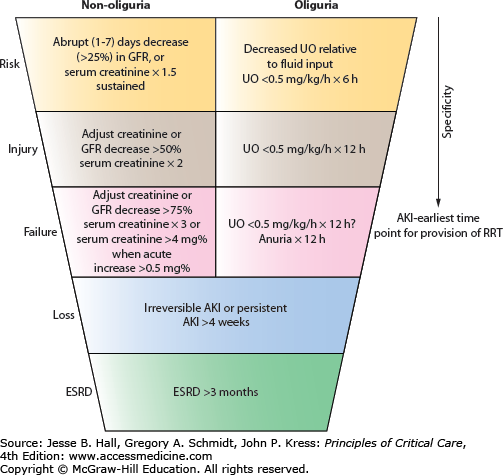
A modification of the RIFLE classification was introduced by the Acute Kidney Injury Network (AKIN) in 2007.6 It differs from RIFLE in a number of ways: it introduces an increase in serum creatinine by 0.3 mg/dL (≥26.4µmol/L) into the definition of AKI; uses changes in serum creatinine within a time window of 48 hours to define AKI, instead of referring to a baseline value; and the requirement of RRT is taken into consideration for staging. AKI according to AKIN is defined as an abrupt (within 48 hours) reduction in kidney function currently defined as an absolute increase in serum creatinine of more than or equal to 0.3 mg/dL (≥26.4µmol/L), a percentage increase in serum creatinine of more than or equal to 50% (1.5-fold from baseline), or a reduction in urine output (documented oliguria of less than 0.5 mL/kg per hour for >6 hours). Staging of AKI according to AKIN classification is detailed in Table 97-1. Staging of AKI is important because the risk for death and RRT rises with increased stage of AKI. Patients should be staged according to the criteria that give them the highest stage, when staging based on creatinine and urine output criteria differ. The Kidney Disease: Improving Global Outcomes (KDIGO) group published clinical practice guidelines for acute kidney injury in 2012.7 These guidelines acknowledged that two validated classification systems existed, but recognized the need for a single definition of AKI for research and clinical practice. KDIGO defines AKI as an increase in SCr by ≥0.3 mg/dL (≥26.5μmol/L) within 48 hours; or an increase in SCr to ≥1.5 times baseline, which is known or presumed to have occurred within the prior 7 days; or a urine volume of ≤0.5 mL/kg per hour for 6 hours or longer. KDIGO uses a staging system similar to AKIN.
Classification/Staging System for Acute Kidney Injurya
| Stage | Serum Creatinine Criteria | Urine Output Criteria |
|---|---|---|
| 1 | Increase in serum creatinine of more than or equal to 0.3 mg/dL (≥26.4μmol/L) or increase to more than or equal to 150% to 200% (1.5- to 2-fold) from baseline | Less than 0.5 mL/kg per hour for more than 6 hours |
| 2b | Increase in serum creatinine to more than 200% to 300% (>2- to 3-fold) from baseline | Less than 0.5 mL/kg per hour for more than 12 hours |
| 3c | Increase in serum creatinine to more than 300% (>3-fold) from baseline (or serum creatinine of more than or equal to 4.0 mg/dL (≥354μmol/L) with an acute increase of at least 0.5 mg/dL (44μmol/L) | Less than 0.3 mL/kg per hour for 24 hours or anuria for 12 hours |
The incidence of AKI varies with the clinical setting and definition used. AKI occurs in 2% to 5% of general medical-surgical admissions, but up to 10% to 30% of ICU admissions. Acute kidney injury can be classified into three broad etiologic/anatomic categories: prerenal AKI, intrarenal AKI, and postrenal AKI. Prerenal AKI is caused by renal hypoperfusion with intact renal parenchyma (55%-60% of instances of AKI). Intrarenal AKI is caused by parenchymal renal diseases (35%-40% of instances of AKI). Postrenal AKI is caused by urinary tract obstruction (∼5% of instances of AKI).8
Most AKI in the ICU is caused by prerenal azotemia, and the rest predominantly by renal parenchymal injury with acute tubular necrosis (ATN); the former may convert to the latter. Despite advances in critical care and dialysis technologies, the mortality of AKI requiring acute renal replacement therapy (RRT) in critically ill patients remains 50% to 80%.9-11 Outcome is particularly poor in septic ARF; in one study, mortality of septic ARF was 74%, compared to 45% in nonseptic cases.12 ARF in the presence of shock (septic or cardiogenic) is an increasingly common occurrence in modern ICUs, and has driven the development of continuous renal replacement therapy (CRRT) to permit control of azotemia and fluid balance in hemodynamically unstable patients.11,13
Although there is evidence that isolated AKI itself increases mortality, it is clear that mortality of ICU AKI increases with every additional nonrenal organ failure present at the time of initiation of renal replacement therapy (RRT).9,11 Emerging data suggest that renal ischemia-reperfusion injury and the uremic milieu actually contribute to the development of distant organ injury (increased pulmonary vascular permeability and cardiac and splanchnic organ apoptosis).14-16 Evidence from animal models suggests that this interaction arises in part from systemic inflammatory changes, activation of proapoptotic pathways, increases in leukocyte trafficking and dysregulated aquaporin channel expression. Identification and modulation of these pathways will be critical to efforts aimed at improving outcomes in patients with AKI.17
AKI with non-recovery can cause end stage renal disease (ESRD) directly; usually when superimposed on significant chronic kidney disease (CKD), or, more rarely due to bilateral renal cortical necrosis. Typically, however, AKI episodes are followed by renal tubular regeneration and apparent recovery. However, in recent years, epidemiologic evidence has accumulated that a significant subset of ESRD is caused by AKI, including de novo cases that are not superimposed on preexisting CKD. Experimental models have demonstrated persistent alterations in kidney structure and function following renal tubular injury, including reduced renal mass, vascular insufficiency, cell cycle disruption (arrest), and maladaptive, fibrosing repair.18 A meta-analysis of 48 studies involving 47,017 patients who were followed up for at least 6 months examined the incidence of CKD, cardiovascular disease and mortality following one episode of AKI.19 Fifteen studies provided follow-up mortality data on controls. The mortality rate in these studies was 8.9 deaths/100 person-years for patients with AKI who survived hospitalization compared with 4.3 deaths/100 person-years for patients who survived hospitalization without AKI (relative risk [RR], 2.59; 95% confidence interval [CI], 1.99-3.42). Cardiovascular outcomes following AKI were examined in 2 studies. Approximately 15.4% of survivors of AKI and 7.0% of survivors without AKI had an MI at 1 year post AKI (RR 2.05; 95% CI, 1.61-2.61). The rate of CKD after AKI was 7.8 events/100 patient-years, and the rate of ESRD was 4.9 events/100 patient-years.19 Of course, the long-term risk of cardiovascular disease is markedly increased by the development or worsening of chronic kidney disease after AKI. In addition, the severity and frequency of AKI episodes are important predictors of poorer outcomes. The KDIGO clinical practice guidelines recommend clinical follow-up of patients 3 months after developing AKI, to determine whether new or worsening CKD has developed, to guide further renal and cardiovascular risk management.7
Although a broad differential diagnosis and therapeutic plan should be considered for every ICU patient with AKI, in most cases the approach is to consider the possibility of urinary tract obstruction, reverse any element of prerenal azotemia, provide supportive care in the presence of ATN, and intervene with effective RRT when indicated, in anticipation of probable renal recovery in the event of patient survival.
CLASSIFICATION OF ACUTE RENAL FAILURE
Prerenal azotemia is the most common cause of AKI in hospitalized patients. The main features of prerenal AKI are decreased renal perfusion (often in the setting of decreased systemic perfusion), the presence of intact renal parenchymal tissue, and the rapid correction of GFR with restoration of renal perfusion. Uncorrected and/or severe prerenal azotemia predisposes to the development of ischemic ATN. Prerenal AKI is caused by any condition leading to renal hypoperfusion, including systemic hypoperfusion with hypovolemia, cardiac failure, or vasodilatory shock, and/or regional hypoperfusion caused by renal vasoconstriction (Table 97-2).
Causes of Acute Renal Failure
| Prerenal |
| Volume depletion: Gastrointestinal fluid loss or hemorrhage; renal losses (diuretics or glucosuria, salt-wasting nephropathy, diabetes insipidus, or adrenal insufficiency); cutaneous losses (burns, desquamation) |
| Volume redistribution: Peripheral vasodilation (sepsis or antihypertensives), peritonitis, burns, pancreatitis, hypoalbuminemia (nephrotic syndrome or hepatic disease) |
| Cardiac dysfunction: Pericardial tamponade, complications of myocardial infarction, acute or chronic valvular disease, cardiomyopathies, arrhythmias |
| Vasodilatory shock: Sepsis, liver failure, postcardiotomy, anaphylaxis, or antihypertensives |
| Renal vasoconstriction: Cirrhosis, sepsis, hypercalcemia, drugs (cyclosporine, tacrolimus, nonsteroidal anti-inflammatory drugs, or pressors) |
| Renal |
| Ischemia: Trauma, surgery, sepsis, pigment nephropathy (hemolysis or rhabdomyolysis), cardiac or aortic hemorrhage |
| Nephrotoxic: Radiocontrast, antibiotics (aminoglycosides or amphotericin), nonsteroidal anti-inflammatory drugs, carbon tetrachloride, ethylene glycol, heavy metals (lead, mercury, arsenic, cadmium, or uranium), pesticides, fungicides, cyclosporine, or tacrolimus |
| Disorders of glomeruli and blood vessels: Poststreptococcal glomerulonephritis, infective endocarditis, systemic lupus erythematosus, Goodpasture syndrome, microscopic polyarteritis, Wegener granulomatosis, Henoch-Schöunlein purpura, idiopathic rapidly progressive glomerulonephritis, polyarteritis nodosa, malignant hypertension, thrombotic microangiopathies (hemolytic uremic syndrome, thrombotic thrombocytopenic purpura, postpartum renal failure, or antiphospholipid syndrome), renal artery embolism, renal artery dissection, bilateral renal vein thrombosis, or abdominal compartment syndrome |
| Acute interstitial nephritis |
| Allergic: Semisynthetic penicillin analogues (eg, methicillin, ampicillin, or nafcillin), cephalosporins, rifampin, ciprofloxacin, cotrimoxazole, sulfonamides, thiazides, furosemide, allopurinol, phenytoin, tetracyclines, or warfarin |
| Infectious: Streptococcal, staphylococcal, leptospirosis, infectious mononucleosis, diphtheria, brucellosis, Legionnaire disease, toxoplasmosis, or cytomegalovirus |
| Infiltrative: Sarcoidosis, lymphoma, leukemia |
| Autoimmune/alloimmune: Systemic lupus erythematosus or renal transplant rejection |
| Postrenal |
| Malignancy: Lymphoma, renal adenocarcinoma, bladder ureteral carcinoma, gynecologic cancers, prostate cancer, other pelvic tumors, or metastatic disease |
| Inflammatory processes: Tuberculosis, inflammatory bowel disease, retroperitoneal abscess or fibrosis, postradiation therapy |
| Vascular diseases: Aortic aneurysm, renal artery aneurysm |
| Papillary necrosis: Diabetes mellitus, sickle hemoglobinopathy, analgesic abuse, prostaglandin inhibition, or hepatic cirrhosis |
| Intratubular: Uric acid, calcium phosphate, Bence Jones proteins, methotrexate, acyclovir, sulfonamide antibiotics, or indinavir |
| Miscellaneous: Nephrolithiasis, ureteral ligation, retrograde pyelography with ureteral edema, neurogenic bladder, neuropathic ureteral dysfunction, or obstructed urinary catheter |
Renal blood flow and GFR are relatively maintained during mild hypoperfusion, due to compensatory mechanisms.20 Renal perfusion is largely preserved within a range of mean arterial pressure (MAP) between 80 and 180 mm Hg, if cardiac output is adequate. As MAP falls below 80 mm Hg, there is a precipitous fall in renal blood flow and GFR.21 There are two major mechanisms of renal blood flow autoregulation: a myogenic reflex and tubuloglomerular (TG) feedback. The myogenic reflex is mediated by stretch receptors in the afferent arterioles, which detect a decrease in perfusion pressure, leading to autoregulatory relaxation of afferent arterioles and vasodilation. TG feedback defends renal perfusion as follows: chloride concentration is continuously sensed in the tubular lumen by the macula densa, just distal to the thick ascending loop of Henle. When luminal chloride decreases (presumably reflecting decreased renal blood flow, GFR, intravascular volume, or a combination of these), a vasodilatory signal is transduced to the corresponding afferent arteriole (and vice versa if flow increases). Together, these mechanisms autoregulate renal blood flow in the face of hypotension or hypertension. A third mechanism additionally helps autoregulate GFR. Increased renin secretion stimulated by hypotension/hypovolemia sensed in the afferent arteriole helps maintain GFR (but not renal blood flow) during hypotension, through the efferent arteriolar action of angiotensin II (discussed further below).
Of course, in addition to local autoregulation, systemic neurohormonal influences also play a prominent role in determining the renal response to shock. Systemic hypoperfusion activates the sympathetic nervous system and renin-angiotensin-aldosterone axis. Norepinephrine and angiotensin II are systemic vasoconstrictors, and tend to increase renal blood flow by preserving renal perfusion pressure. On the other hand, both hormones are renal vasoconstrictors, though they differ in their glomerular hemodynamic effects. Angiotensin II preferentially constricts efferent arterioles, and helps preserve glomerular filtration, increasing filtration fraction (the ratio of GFR to renal plasma flow) by creating “back-pressure” to augment net filtration pressure in the glomerular capillary. Norepinephrine causes balanced afferent and efferent arteriolar constriction, similarly increasing filtration fraction in the face of decreased renal blood flow, but to a lesser extent than angiotensin II. Both angiotensin II and norepinephrine stimulate intrarenal vasodilator prostaglandin production, thus attenuating their simultaneous effect of afferent arteriolar vasoconstriction and helping preserve renal perfusion.
Drugs may unfavorably alter the glomerular hemodynamic response to renal hypoperfusion. Nonsteroidal anti-inflammatory drug (NSAID) administration to patients with decreased effective arterial blood volume (due to hypovolemia or congestive heart failure) or renal vasoconstriction (due to cirrhosis) leads to a decline in renal blood flow and GFR. These patients are dependent on vasodilator prostaglandins to maintain renal perfusion, so NSAIDs leave renal vasoconstrictor influences unopposed. Angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) can lead to prerenal azotemia in patients who are dependent on angiotensin II for maintenance of GFR. This phenomenon is most commonly seen in patients receiving ACEIs or ARBs in the presence of hypovolemia, bilateral renal artery stenosis, or unilateral renal artery stenosis with a solitary kidney.
Prerenal azotemia also leads to avid renal tubular sodium and water reabsorption throughout the nephron. Catecholamines and angiotensin II directly increase sodium transport and reabsorption in the proximal and distal nephron. Efferent arteriolar constriction by angiotensin II and increased filtration fraction simultaneously lead to decreased peritubular capillary hydraulic pressure and increased peritubular capillary oncotic pressure. The combination of high oncotic pressure and low hydraulic pressure in peritubular capillaries increases sodium and water absorption in the proximal tubule, a process termed glomerulotubular balance. Angiotensin II also leads to downstream production of aldosterone, another salt-retaining influence. Severe hypovolemia/hypotension (>10%-15% decrease in MAP or blood volume) leads to nonosmotic vasopressin secretion, and avid water reabsorption in the collecting duct, along with systemic vasoconstriction. Finally, in hypovolemic patients, decreased atrial stretch downregulates production of atrial natriuretic peptide, also favoring sodium retention (the opposite is true if renal hypoperfusion is caused by congestive heart failure). Thus the combination of glomerulotubular balance and the tubular effects of catecholamines, angiotensin II, aldosterone, and vasopressin mediate the salt and water retention which is the hallmark of prerenal azotemia. Accordingly, patients with prerenal azotemia tend to have oliguria, low urine sodium, and concentrated urine with a urine osmolality exceeding 500 mOsm/kg. Low urine sodium (and fractional excretion of sodium; see below) and increased urine osmolality (with a high urine:plasma creatinine ratio) are not seen in patients who have prerenal AKI due to renal losses (ongoing diuretic therapy, salt-wasting nephropathies, osmotic diuresis, adrenal insufficiency, and central or nephrogenic diabetes insipidus). Other common laboratory features of prerenal AKI are increased serum BUN:creatinine ratio (caused by low tubular flow and increased urea reabsorption), decreased fractional excretion of urea (see below), polycythemia/high serum albumin (hemoconcentration), mild hypercalcemia, hyperuricemia, and acid-base abnormalities (metabolic acidosis from diarrhea or shock or lactic acidosis; metabolic alkalosis from diuretics or vomiting). Hyponatremia may also be present, depending on abnormalities in water balance (see Chap. 99). The renal response to volume challenge or vasoactive drug initiation may also be used to determine the presence or absence of a reversible, “prerenal” etiology of AKI.
Postrenal AKI (often called obstructive uropathy) accounts for 5% of all cases of AKI, and is more common in the elderly. Unilateral obstruction is not sufficient alone to cause AKI. Renal insufficiency due to obstruction occurs only when the obstruction involves a site that affects both kidneys, or a single functioning kidney. The most common cause of postrenal obstruction is bladder neck obstruction (prostatic hypertrophy, prostate cancer, and neurogenic bladder). Postrenal obstruction is also caused by bilateral ureteral obstruction or unilateral obstruction in patients with solitary kidney (stones, clots, sloughed renal papillae, retroperitoneal fibrosis, or retroperitoneal masses). Intratubular obstruction can be caused by crystals like uric acid, calcium oxalate, calcium phosphate, acyclovir, sulfadiazine, indinavir, methotrexate, or by paraprotein (myeloma cast nephropathy). Volume expansion, sometimes with urinary alkalinization (uric acid, methotrexate, or myeloma), is the primary treatment for these causes of intratubular obstruction.
Obstruction to urine flow by increased tubular hydrostatic pressure is only partly responsible for the reduced GFR of obstructive uropathy. AKI is also caused and sustained by renal vasoconstriction that occurs in response to ureteral obstruction, mediated by thromboxanes. Obstruction should be suspected in patients with recurrent urinary tract infections, nephrolithiasis, prostate disease, or pelvic tumor. Causes of obstructive uropathy are listed in Table 97-2. These patients usually have a preceding history of obstructive symptoms followed by sudden onset of anuria or oliguria. Polyuria and nocturia due to renal concentrating defect may be seen in patients with partial or intermittent obstruction. Other features of AKI secondary to obstruction are increased BUN:creatinine ratio, hyperkalemia, and defective urinary acidification with metabolic acidosis.
The two most important diagnostic tests when obstruction is suspected are bladder catheterization and renal ultrasonography. If urinary tract obstruction is strongly suspected, but ultrasound results are equivocal, then a “stone protocol” noninfused computed tomography (CT) scan should be performed. In some cases where false-negative ultrasound or CT scan results are suspected, cystoscopy and retrograde pyelograms may be required to definitively exclude the diagnosis of obstructive uropathy. For example, we would request retrograde pyelograms despite normal ultrasound images in a patient with anuric, hyperkalemic AKI and extensive pelvic tumor, potentially encasing the ureters and preventing dilation and hydronephrosis. Retroperitoneal fibrosis can similarly cause obstructive AKI without hydronephrosis. Early diagnosis is essential, as the extent of parenchymal damage is dependent on the duration of obstruction; complete recovery is possible up until 10 to 14 days of obstruction.
Intrinsic acute renal failure can be categorized anatomically, according to the site of the lesion: vascular, glomerular, or tubulointerstitial. We will discuss tubular and interstitial causes first, because they are far more common in hospitalized patients.
Acute Tubular Necrosis: The most common cause of intrinsic AKI in hospitalized patients is acute tubular necrosis (ATN). ATN is caused by ischemia, nephrotoxins, or a combination of both, and accounts for approximately 85% to 90% of intrinsic AKI cases.8,22 Ischemic ATN is commonly seen in patients with sepsis or severe cardiac failure, or postoperative patients, particularly after cardiac and aortic surgeries. Massive trauma or cardiac arrest are other causes of ATN. Prerenal failure can result in ischemic ATN if renal hypoperfusion is severe and not reversed by timely therapy. Although improving renal perfusion may reverse prerenal AKI (by definition), and diminish ischemic contributions to the pathogenesis of ATN, it is quite conceivable that in many cases ATN develops despite appropriate resuscitation and adequate renal perfusion. Zager has shown in an endotoxemic rat model of septic AKI that paired combinations of insults (renal cross clamp, systemic endotoxin, amino-glycoside, and temperature elevation) cause azotemia and renal pathologic findings of ATN, but these insults individually cause no renal dysfunction or injury.22 We suspect that this synergistic injury model accurately reflects the pathogenesis of much AKI in the ICU. Positive pressure mechanical ventilation alters renal perfusion and function through a variety of mechanisms, both hemodynamic and inflammatory.23 Other experimental data have shown that endotoxin, tumor necrosis factor, and numerous other inflammatory mediators are directly cytotoxic to renal endothelial and tubular cells.24
The diagnosis of ATN is usually made by clinical exclusion of alternative diagnoses such as obstruction or prerenal azotemia, and a lack of features suggestive of other intrinsic renal lesions. Characteristic urinalysis and urine chemistry findings frequently support the clinical diagnosis, increasingly combined with analysis of novel biomarkers of renal tubular damage (although the latter are not currently available for clinical use in the USA). Ischemic ATN is usually reversible by tubular regeneration in surviving patients with previously normal renal function; although, as discussed above, in a significant subset of cases, recovery may be less complete than is clinically apparent. Failure to recover prompts consideration of a differential diagnostic list including bilateral cortical necrosis, renal atheroembolism, renal artery stenosis/thrombosis/dissection, and severe forms of other intrinsic lesions such as rapidly progressive glomerulonephritis (GN). Bilateral cortical necrosis causes irreversible renal failure and is associated with profound shock with disseminated intravascular coagulation, obstetric complications, hemolytic uremic syndrome, or snake bites.25
Cellular mediators that play a role in the pathogenesis of ATN include calcium, reactive oxygen species, phospholipases, proteases, adhesion molecules, and nitric oxide (NO).26 ATN has several phases: prerenal, initiation, extension, maintenance, and repair.27 It is not intuitive that renal tubular injury should cause decreased glomerular filtration and AKI. A decrease in glomerular ultrafiltration coefficient has been shown in several animal models of AKI, but this is a minor contributor to the observed decrement in GFR.28,29 The pathophysiologic mechanisms that explain the reduction of GFR in ATN are hemodynamic abnormalities, tubular obstruction, and tubular back leakage of glomerular filtrate (Fig. 97-2).30 Renal vasoconstriction is seen in AKI,30,31 caused by activation of tubuloglomerular feedback; increased distal chloride delivery past injured tubular segments is sensed by the macula densa, causing vasoconstriction of the corresponding afferent arteriole. This reversible, functional mechanism seems to be the major cause of decreased GFR in ATN, and is in part protective. Severe hypovolemia would rapidly result if injured tubules failed to reabsorb the bulk of filtered sodium and water; thus the term “acute renal success” has been used to describe the development of decreased GFR (“acute renal failure”) in the presence of tubular necrosis.31 Furthermore, reabsorption of filtered sodium accounts for the bulk of renal oxygen consumption; continued glomerular filtration of sodium in ATN may aggravate hypoxic damage to sublethally injured tubules. The phenomenon of medullary hypoxia plays an important role in the pathogenesis of ATN. Low medullary blood flow is required for urinary concentration.32 Reabsorption of sodium chloride by the medullary thick ascending limb of the loop of Henle (mTAL) is the major determinant of medullary oxygen consumption, resulting in a hypoxic environment under normal circumstances.32,33 mTAL is vulnerable to ischemic injury if increased oxygen requirement is associated with decreased oxygen delivery. In addition, the inflammatory mechanisms that dominate as ATN progresses from initiation to extension and maintenance phases of ATN result in medullary congestion and hypoperfusion.27
FIGURE 97-2
Pathophysiologic mechanisms of acute kidney injury. Tubular damage by ischemia, nephrotoxins, or both, leads to decreased GFR by a combination of mechanisms. (1) Renal vasoconstriction via activation of tubuloglomerular feedback, and decreased vasodilator substances (PGI2, prostacyclin; NO, nitric oxide) is a prominent functional mechanism of decreased GFR in ATN. (2) Backpressure from tubular obstruction by casts directly decreases GFR. (3) Backleakage of glomerular filtrate into peritubular capillaries decreases the efficiency of glomerular filtration, effectively decreasing GFR. (4) There is increasing evidence for a role of interstitial inflammation in the extension phase of ATN. (5) Direct glomerular effects (mesangial contraction, decreased filtration surface area) may also play a role in decreasing GFR in the presence of ATN. (Reproduced with permission from Lameire N, Vanholder R. Pathophysiologic features and prevention of human and experimental acute tubular necrosis. J Am Soc Nephrol. February 2001;(12 suppl 17):S20-S32.)
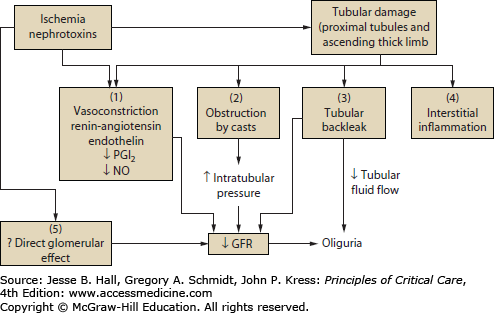
The tubular factors that are also involved in the reduction of GFR in AKI are tubular obstruction and tubular back leakage. Necrotic cell debris incorporated into casts causes obstruction of proximal and distal tubules and has been shown to play a significant role in experimental AKI.34,35 Back leakage of tubular fluid across denuded basement membranes and injured proximal tubule cells has been demonstrated in several experimental models of AKI.36 Subsequently it has been shown that tubular back leakage and intratubular obstruction are important factors contributing to the reduction of GFR in human ischemic AKI.37,38
Nephrotoxic injury is the second major cause of ATN. Nephrotoxic ATN is caused by drugs (aminoglycosides, cisplatin, amphotericin, and chemotherapy), radiocontrast agents, heme pigments (myoglobin and hemoglobin), and myeloma light chain proteins. AKI due to aminoglycosides and radiocontrast agents accounts for most cases of nephrotoxic ATN. Nephrotoxicity of cancer chemotherapy is discussed in Chap. 95; malignancy and AKI are further discussed below. Three nephrotoxic AKI syndromes will be further discussed here: aminoglycoside nephrotoxicity, radiocontrast nephropathy, and AKI caused by nonsteroidal anti-inflammatory drugs (NSAIDs).
Aminoglycoside Nephrotoxicity Aminoglycosides like tobramycin, gentamicin, amikacin, and netilmicin are widely used for the treatment of gram-negative infections. Although they are effective antibiotics, therapy with aminoglycosides is complicated by nephrotoxicity in 10% to 20% of patients.39 Aminoglycosides are excreted by glomerular filtration and are reabsorbed by proximal tubular cells. The mechanism of aminoglycoside-induced renal injury is incompletely understood. Accumulation of aminoglycosides in the proximal tubules in high concentration results in disruption of a variety of intracellular processes. Aminoglycosides are tubular toxins, and the earliest morphologic changes consist of vacuolization of proximal tubules, loss of brush border, and the presence of myeloid bodies within proximal tubule cells. Clinical evidence also attests to the tubular toxicity of aminoglycosides; maximum urine osmolality falls, and renal wasting ofmg2+ and K+ ensues. The relationship between this tubule damage and reduced GFR remains unclear, although in experimental models using high doses of aminoglycosides, tubule obstruction and back leakage can be demonstrated. In experimental models of aminoglycoside nephrotoxicity, relatively small doses decrease glomerular permeability, while larger doses cause renal vasoconstriction. The relevance of these hemodynamic changes to human aminoglycoside nephrotoxicity is not known.
AKI generally develops 7 to 10 days after aminoglycoside therapy is started. Aminoglycoside nephrotoxicity is associated with nonoliguric AKI, due to a concentrating defect caused by tubular injury. Aminoglycoside nephrotoxicity is also accompanied by potassium and magnesium wasting. Risk factors for aminoglycoside nephrotoxicity are summarized in Table 97-3. In patients with preexisting renal insufficiency, the dosing interval should be adjusted and levels monitored to minimize the risk of ototoxicity and nephrotoxicity. Once-daily dosing of aminoglycosides decreases nephrotoxicity with no apparent loss of effectiveness.40-42 In patients with acutely deteriorating renal function who require aminoglycoside therapy, monitoring may be very difficult and require frequent reassessment. Aminoglycosides should ideally be discontinued whenever renal dysfunction develops; increasing trough levels and decreased calculated aminoglycoside clearance may signal decreased GFR before serum creatinine increases. Recovery of renal function is expected in most cases when nephrotoxicity is recognized early and aminoglycosides held, but may be delayed for days to weeks.
Contrast-Induced AKI AKI due to radiocontrast agents occurs within 24 to 48 hours of intravenous radiocontrast administration, and is termed contrast-induced (CI-AKI). It has most commonly been defined in the literature as a rise in serum creatinine of ≥0.5 mg/dL (≥44μmol/L) or a 25% increase from baseline value, assessed at 48 hours after a radiological procedure; but recent studies typically use the RIFLE, AKIN, or KDIGO definitions for case definitions.43 Vasoconstriction and direct tubular toxicity due to generation of oxygen free radicals are thought to be the pathogenic mechanisms of radiocontrast nephrotoxicity. In most cases, the AKI is mild and recovery typically begins with stabilization of serum creatinine at 3 to 5 days postcontrast.44 Renal failure is usually nonoliguric. However, in patients with preexisting severe chronic renal failure, CI-AKI may be severe, irreversible, and require chronic dialysis. Patients who develop CI-AKI are at increased risk of death or prolonged hospitalization, as well as for other adverse outcomes, including early or late cardiovascular events. The mortality is higher in patients who develop severe CI-AKI requiring dialysis compared to those not requiring dialysis. In a study of 1896 patients undergoing coronary angiography, the hospital mortality was 7.1% in CI-AKI and 35.7% in patients who required dialysis.45
The risk of nephrotoxicity due to contrast agents varies with the type and dose of agent. Low- or iso-osmolal agents are less nephrotoxic than ionic high-osmolal agents.46,47 The incidence of AKI due to radiocontrast agents is less than 2% in patients with normal renal function, but is inversely related to GFR in patients with CKD; in high-risk patients the incidence is as high as 60%.48 There are several important risk factors for radiocontrast nephropathy, including CKD (the major risk factor; CI-AKI is usually a form of AKI superimposed on CKD), diabetic nephropathy (see below), severe congestive heart failure, intravascular volume depletion, high contrast dose, and multiple myeloma (Table 97-4). Of note, the risk of radiocontrast nephropathy at any GFR level in CKD is approximately double in diabetics compared to nondiabetics. The group at risk for radiocontrast nephropathy can be reliably identified, and a variety of prophylactic strategies have been used, several successfully.
A number of measures are used for prevention of radio-contrast nephropathy. Alternative imaging approaches (ultrasound, noninfused CT) are preferred to radiocontrast studies in patients with a high risk of radiocontrast nephropathy. Nonionic low-osmolal agents46 and nonionic iso-osmolol agents47 are less nephrotoxic, and these agents are used to decrease the incidence of AKI due to radiocontrast agents. Volume expansion has been shown to reduce the incidence of CIN in a number of studies, but the optimum solution and administration regimen has yet to be conclusively demonstrated. Isotonic saline (1 mL/kg per hour for 12 hours pre- and postcontrast) was associated with a lower incidence of CIN than a hypotonic saline regimen in a prospective randomized trial involving 1620 patients.49 It has been suggested that sodium bicarbonate may be superior to normal saline, as urinary alkalinization may reduce free radical formation. The results of randomized controlled trials and meta-analyses have been variable with some reporting lower rates of CIN and others equivalent rates. The largest meta-analysis published involved 14 trials and 2290 patients, but there was considerable clinical and statistical heterogeneity between trials (largely owing to variable trial size). Pooling the three large trials (1145 patients), there was no difference between CI-AKI rates with sodium chloride or sodium bicarbonate. The pooled relative risk of CI-AKI with sodium bicarbonate was 0.50 (0.27-0.93) in the 12 smaller trials; however, these trials were of lower methodological quality.50
In some studies, the administration of acetylcysteine, a thiol-containing antioxidant, in combination with saline hydration has been shown to be beneficial in reducing the incidence of contrast nephropathy when administered in various oral regimens (1200 mg once or 600 mg every 12 hours before and after radiocontrast).51-53 One successful study used intravenous acetylcysteine for radiocontrast nephropathy prophylaxis.54 Although other studies did not show any benefit,55 a recent meta-analysis of eight randomized controlled trials involving 855 patients reported that the use of acetylcysteine reduced the risk of radiocontrast by 59%.56 However, further doubt has been cast over these inconsistent findings by emerging experimental data that suggest that the apparent efficacy of acetylcysteine in these studies may have been artifactual. Specifically, acetylcysteine causes a decrement in serum creatinine (but not cystatin C) by a GFR-independent mechanism,57 perhaps by inhibiting creatinine phosphokinase function.58 In one of the largest randomized studies to date, involving 2308 patients assessing the utility of oral N-acetylcysteine to prevent CIN, the authors concluded that it did not lower the risk of CI-AKI or other renal outcomes.59 However, the generalizability of this study may be limited; only 362 patients (15.7%) had a baseline serum creatinine greater than 1.5 mg/dL, the median contrast volume was low at 100 mL and there was imprecision regarding which patients received hydration and what volume was given. It may be more appropriate to conclude that N-acetylcysteine is not effective at preventing CI-AKI in low-risk groups; however, it may still have a role in preventing CI-AKI in patients at high risk.
Other approaches such as vasodilation with dopamine60 or fenoldopam61 failed to prevent radiocontrast nephropathy, and mannitol and furosemide appeared to increase the rate of AKI postcontrast in one study.62 Prophylactic hemodialysis has not been shown to be beneficial in the prevention of contrast nephropathy.63 A recent meta-analysis looked at nine randomized and two nonrandomized trials involving 1010 patients who underwent RRT (hemodialysis or hemofiltration) to prevent CI-AKI. RRT did not decrease CI-AKI incidence compared with standard medical therapy (RR 1.02; 95% CI, 0.54-1.93). However, intertrial heterogeneity was high. Limiting the analysis to trials involving hemodialysis only (eight trials) reduced heterogeneity. Hemodialysis appeared to confer an increased risk of CI-AKI (RR 1.61; 95% CI, 1.13-2.28) and had no effect on need for permanent RRT or progression to ESRD (RR 1.47; 95% CI, 0.56-3.89).64 Since RRT is also expensive, associated with a variety of risks, and a limited resource, we do not recommend this approach to CI-AKI prevention.
A novel method of preventing CI-AKI is the RenalGuard system (PLC Medical, Franklin, MA); a fluid management device which precisely measures urine output and replaces the same amount of fluid intravenously. The REMEDIAL II trial evaluated the effects of a forced diuresis induced by furosemide and replacement with saline as guided by the RenalGuard system.65 This strategy theoretically would reduce contrast exposure to tubular cells and increase its elimination. It evaluated the incidence of CI-AKI in 392 high-risk patients with an eGFR ≤30 mL/min. Patients randomized to the RenalGuard system achieved urinary flow rates of ∼350 mL/min. CI-AKI occurred in 30/146 (20.5%) of patient in the control group (N-acetylcysteine and isotonic bicarbonate solution) compared with 16/146 (11%) in the RenalGuard group (N-acetylcysteine, furosemide, and isotonic saline administration as controlled by the RenalGuard system), (OR, 0.47; 95% CI 0.24-0.92).65 However, concerns have been raised about the volume of fluid replacement precipitating pulmonary edema in patients with cardiac dysfunction, and the development of electrolyte abnormalities such as hypokalemia developing following diuretic use. Further research is required to determine the optimal approach to CI-AKI prevention.
Nonsteroidal Anti-Inflammatory Drugs and Acute Renal Failure NSAIDs can cause hemodynamically mediated AKI. Vasodilatory prostaglandins (prostacyclin and prostaglandin E2 [PGE2]) are essential for the maintenance of renal blood flow and GFR in states of effective volume depletion, such as congestive heart failure, cirrhosis of the liver, nephrotic syndrome, and in states of true volume depletion.66 Prostaglandins counterbalance the effects of vasoconstrictors such as angiotensin II and catecholamines. NSAIDs inhibit prostaglandins and thus would lead to unopposed effect of angiotensin II and catecholamines, leading to reduced GFR. Hyperkalemia may be prominent because NSAIDs impair renin secretion. Patients with CKD are similarly at risk for NSAID-induced AKI, because vasodilator prostaglandins maintain hyperfiltration in remnant nephrons. Hemodynamically mediated AKI is seen within days of taking NSAIDs in high-risk patients. Cyclooxygenase-2 (COX-2) selective drugs have the same renal effects as nonselective NSAIDs, because COX-2 is constitutively expressed and physiologically active in the kidney.67 The second type of acute renal injury is allergic interstitial nephritis, as discussed below. Chronic NSAID use is also associated with papillary necrosis and is thought to occur in patients who are taking multiple analgesics.
Acute Tubulointerstitial Nephritis: AKI due to acute interstitial nephritis (AIN) is most often caused by allergic reaction to various drugs (allergic AIN), but there are also a variety of infectious (Legionnaire disease, cytomegalovirus, and Hantavirus), autoimmune (lupus), alloimmune (renal transplant rejection), and infiltrative (sarcoidosis, leukemia, and lymphoma) disorders that can cause AIN (see Table 97-2). The most common drugs that cause allergic AIN are penicillins, cephalosporins, ciprofloxacin, rifampin, sulfonamides (furosemide, thiazide diuretics, and trimethoprim-sulfamethoxazole), proton pump inhibitors (increasingly recognized as a cause of AIN), H2-blockers, allopurinol, and NSAIDs.
Patients with AIN classically present with AKI temporally related to drug therapy or infection, associated with a triad of fever, rash, and eosinophilia.68 Urinalysis findings include leukocyturia with eosinophiluria, leukocyte casts, and low-grade proteinuria. Although all these signs are present in the majority of patients with AIN, absence of these does not exclude the diagnosis of AIN. In particular, these findings are usually absent in NSAID-induced AIN. Proteinuria is mild to moderate except in NSAID-induced AIN, in which nephrotic syndrome caused by a minimal change lesion has been described. Tubular dysfunction (eg, Fanconi syndrome, distal renal tubular acidosis, or hyperkalemia) occurs in the majority of patients with AIN.
AIN is usually suspected from the history and laboratory findings. In the absence of urinary tract infection, detection of large numbers of urinary eosinophils (>5%) strongly suggests the diagnosis of drug-induced tubulointerstitial nephropathy (TIN). Hansel stain of the urine is more sensitive than Wright stain for the detection of urinary eosinophils.69 The Hansel method correctly identified 10 of 11 patients with TIN, as opposed to only 2 of 11 correctly classified using Wright stain. False-positive results with the Hansel technique are most commonly caused by rapidly progressive GN or acute prostatitis. Diagnosis can be confirmed by renal biopsy if AKI is progressive and treatment of an alternative diagnosis is considered (eg, rapidly progressive GN), or if there is no recovery of renal function after discontinuation of the medication suspected to have caused AIN.
No therapy is recommended in patients with mild renal insufficiency, and in patients who respond after discontinuation of the offending medication. AIN is usually reversible after withdrawal of the offending agent and treatment of underlying disease. The optimal therapy of AIN is unknown, since there are no randomized controlled trials or large observational studies. Corticosteroid therapy is unproven, and generally recommended only in patients with biopsy-confirmed acute allergic interstitial nephritis who do not respond to conservative management, and have no contraindication to immunosuppression.
Rapidly Progressive Glomerulonephritis: Glomerulonephritis such as postinfectious glomerulonephritis, lupus nephritis, and Goodpasture syndrome can cause acute or subacute renal failure (see Table 97-2). Rapidly progressive glomerulonephritis (RPGN) causing AKI is an emergency. The combination of AKI with an “active” urine sediment (heavy proteinuria, hematuria, leukocyturia, and erythrocyte/leukocyte/mixed cellular casts) should prompt urgent evaluation with renal biopsy and serologies. The underlying pathologic lesion is classically necrotizing crescentic glomerulonephritis, treated with high-dose pulse corticosteroid therapy to stabilize renal function, followed by intensive immunosuppression (with or without plasmapheresis). Causes include inflammatory disorders with immune complex deposition (lupus, postinfectious, or cryoglobulinemia), anti–glomerular basement membrane antibodies (Goodpasture syndrome, when associated with pulmonary hemorrhage), and pauci-immune glomerulonephritis (usually with small-vessel vasculitis; see below). Associated clinical features such as pulmonary hemorrhage, sinus involvement, leukocytoclastic vasculitis, neuropathy, or other stigmata of autoimmune disease may increase the index of suspicion for this diagnosis, but it must be emphasized that a skilled urinalysis demonstrating an “active” sediment in a patient with AKI may make this diagnosis alone. In fact, the presence of erythrocyte casts is pathognomonic of GN; associated with AKI, this is essentially diagnostic of RPGN.
Vascular causes of AKI are categorized into small-vessel diseases and large-vessel diseases (see Table 97-2). Diseases involving small vessels include microscopic polyarteritis (vasculitis in polyarteritis nodosa and Kawasaki disease involves medium-sized vessels), granulomatosis with polyangiitis (Wegener), mixed cryoglobulinemia, and conditions that are categorized as thrombotic microangiopathies, including thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, scleroderma renal crisis, malignant hypertension, and antiphospholipid antibody syndrome. Large-vessel renal vascular diseases include thromboembolic diseases and renal vein thrombosis. Atheroembolic disease should be considered in patients who develop AKI after instrumentation of the aorta, particularly in patients with known atherosclerotic disease.70 Renal vein thrombosis is usually a complication of nephrotic syndrome, and if bilateral can cause AKI. Abdominal compartment syndrome (see Chap. 114) is present when the intra-abdominal pressure (IAP) reaches 20 to 25 cm H2O, and unless decompressed, irreversible organ failure may result.71 The pathogenesis of oliguria and AKI in abdominal compartment syndrome involves venous compression (decreased venous return and renal vein compression), ureteral compression with obstructive uropathy, and possibly changes in renovascular resistance and intrarenal blood flow distribution. Regardless of the underlying cause, a reduction in urine output with or without azotemia in the presence of a measured intra-abdominal pressure over 15 cm H2O is cause for concern and should prompt intervention.
RENAL FUNCTION MONITORING AND DIAGNOSTIC APPROACH TO ACUTE RENAL FAILURE
Attempts to develop strategies to prevent or treat AKI in the ICU have been hampered by the lack of sensitive real-time methods to monitor renal perfusion and function in the clinical setting.72 There are no direct measures of renal perfusion in clinical use. Of the available research techniques for this purpose, para-aminohippuric acid (PAH) clearance is not a valid method to assess renal plasma flow because renal PAH extraction is impaired in critical illness, after cardiac surgery, postrenal transplantation, and in AKI.73 Clinical indices such as plasma concentrations of urea nitrogen and creatinine, urine output, urine chemistries, and urinalysis may be assessed in combination to monitor renal perfusion and function, but alterations in these markers are insensitive, indirect, and often delayed manifestations of renal hypoperfusion and injury.72 Although relatively insensitive, it should be recognized that progressive elevation of serum creatinine is a specific sign of decreased GFR and diagnostic of AKI. As depicted in Figure 97-3, daily serum creatinine increments of 0.5 to 1 mg/dL (depending on muscle mass, and in the absence of rhabdomyolysis) signal severe depression of GFR.74 In the absence of obstruction or renal hypoperfusion, this is usually diagnostic of ATN.
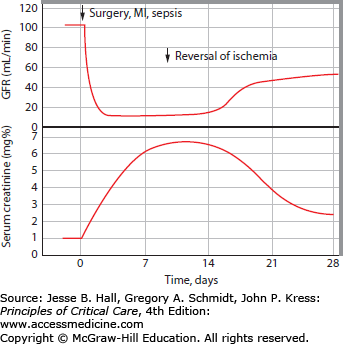
FIGURE 97-3
Dynamic relationship between serum creatinine and GFR in AKI. Moran and Myers demonstrated that elevation of serum creatinine lags significantly behind GFR decrements in AKI. In this example, an acute decrement in GFR to <10 mL/min (commonly a dialysis-requiring level) after a major ischemic insult is associated with a subsequent slow, daily rise of serum creatinine. The daily serum creatinine increment is determined by creatinine generation (muscle mass and catabolic state), volume status (volume of distribution of creatinine), and the new GFR level. Serum creatinine continues to rise until creatinine generation equals creatinine excretion at steady state. Only after a week is it fully apparent from the serum creatinine level how low the GFR is in this patient with AKI; this could have been inferred from the progressive daily serum creatinine increments, and measured by urine collection for an abbreviated creatinine clearance. (Reproduced with permission from Moran SM, Myers BD. Course of acute renal failure studied by a model of creatinine kinetics. Kidney Int. June 1985;27(6):928-937.)
Direct measurement of GFR is probably the most relevant marker of renal function to monitor in the ICU, and can be used as an index of adequate systemic perfusion, a marker of organ dysfunction, a guide to medication dosing, and to determine timing of initiation of renal replacement therapy. GFR measurement methods more sensitive than monitoring serum creatinine and urea nitrogen have been validated in this population, but are not yet in widespread clinical use.72,75 Abbreviated creatinine clearance measurements (2-4 hours) provide a more accurate estimate of GFR than serum creatinine alone, and are more timely than 24-hour urine collections.76 Emerging data suggest that serial monitoring of serum cystatin C levels may provide a more sensitive index of early AKI than serum creatinine or BUN. Cystatin C is a nonglycosylated 13-kDa basic protein that is a member of the cystatin super-family of cysteine protease inhibitors. It is produced by all nucleated cells, and its production rate is unaltered by inflammatory conditions or muscle mass; only thyroid dysfunction has been shown to alter serum levels independent of GFR in studies to date.77-79 It undergoes purely renal excretion. Serum cystatin C levels increase before serum creatinine levels in patients with progressive chronic kidney disease.77,79 Emerging data suggest that plasma cystatin C levels similarly increase 1 to 2 days before serum creatinine in patients developing AKI in a variety of settings,79 including radio-contrast nephropathy,80 renal transplantation,81,82 cirrhotic AKI,83 malarial AKI,84 and AKI in the ICU.85,86 In the latter study of 85 critically ill patients, Herget-Rosenthal and colleagues showed that AKI defined by elevation of cystatin C occurred 1 to 2 days before serum creatinine elevation; the area under the curve in receiver operating curve analysis was 0.82 and 0.97 on the 2 days prior to creatinine-defined AKI.86
In patients with azotemia and/or oliguria, traditional urine chemistry markers may provide useful information, interpreted in combination with other clinical and laboratory parameters (see below), including microscopic urinalysis.72,87 Hopefully, more sensitive markers of early renal hypoperfusion and injury will prove clinically useful in the future.88,89 Meanwhile, all clinically available renal function indices should be used in combination for assessment of renal perfusion and function in the ICU.
The diagnostic approach to AKI involves assignment of the cause to prerenal, renal, or postrenal categories, with further refinement of the diagnosis based on additional laboratory testing.
History and Physical Examination in Acute Renal Failure: Any decrease in effective perfusion of the kidneys can result in the syndrome of prerenal AKI. This may be the result of an absolute decrease in the extracellular fluid (ECF) volume, redistribution of ECF from vascular to interstitial locations (third-spacing), or impaired delivery of blood to the kidneys such as can occur in patients with renal arterial stenosis, vasculitis, or depressed cardiac function. Third-space losses should be suspected in the presence of severe burns, pancreatitis, peritonitis, or recent abdominal surgery. Absolute decreases in the ECF volume are most common in the setting of gastrointestinal fluid losses or in patients receiving excessive doses of diuretics.
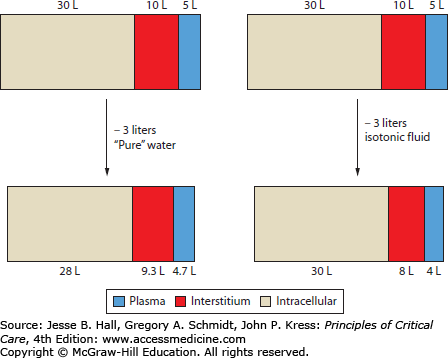
Decreases in weight, if known, can provide some information about the degree of ECF loss. However, weight changes are subject to misinterpretation if the nature of fluid loss is not taken into account (Fig. 97-4). The ability of pure water loss (which is spread out across the total-body water) to cause volume depletion is only one-third as great as an equivalent loss of isotonic fluid (all of which must come from the smaller ECF compartment). Thus the importance of changes in weight should be assessed relative to changes in serum sodium concentration.
FIGURE 97-4
Effect of fluid loss on body water distribution. Because water is in osmotic equilibrium across biologic membranes, loss of 3 L of solute-free water will be spread across the total body water, resulting in a small decrease (0.3 L) in plasma volume. A similar loss of isotonic fluid, which does not obligate osmotic water movement, leads to a much greater decrease (1.0 L) in plasma volume. This is based on the assumption of 45 L of total body water, two-thirds of which is intracellular. Of the extracellular fluid, about one-third is plasma and the remainder extravascular (interstitium).
Related posts:






Full access? Get Clinical Tree


