KEY POINTS
Myocardial ischemia results from an imbalance between myocardial oxygen demand and supply. The major determinants of myocardial oxygen requirements are heart rate, contractility, and wall stress (afterload).
Patients with myocardial ischemia are divided by presentation into those with or without ST elevation, in accordance with treatment strategies. Patients with ST elevation benefit from immediate reperfusion with percutaneous coronary intervention or fibrinolytic agents.
Myocardial infarction is diagnosed by a compatible clinical history, evolution of characteristic ECG changes, and an increase and decrease in cardiac enzymes.
All patients with suspected myocardial ischemia should be given aspirin upon presentation.
Prognosis after myocardial infarction is most closely related to the degree of left ventricular impairment.
Risk stratification is the key to initial management of patients with non-ST elevation acute coronary syndromes.
In patients with high-risk non-ST elevation acute coronary syndromes, an early invasive approach is preferred.
Aspirin, clopidogrel, β-blockers, angiotensin converting enzyme inhibitors, and statins have been shown to decrease mortality after myocardial infarction.
Echocardiography is extremely useful for the diagnosis of complications after myocardial infarction. Invasive hemodynamic monitoring may be necessary in some cases as well.
Patients with cardiogenic shock should be stabilized with an intra-aortic balloon pump and revascularized promptly with percutaneous coronary intervention or bypass surgery.
INTRODUCTION
Myocardial ischemia can go unrecognized in an ICU setting. Signs of myocardial ischemia may be obscured by other illnesses present in the critically ill patient. Physical examination in these patients often is limited, or its results altered, by the presence of other disease processes.
Myocardial ischemia and attendant left ventricular dysfunction may complicate the course and treatment of a particular illness. Conversely, multisystem illness may set the conditions for increased oxygen demand, often accompanied by diminished delivery of oxygen to the heart. For these reasons, the critical care physician must maintain a high index of suspicion for myocardial ischemia in the ICU setting, especially in the patient with a prior history of or multiple risk factors for coronary artery disease.
Myocardial ischemia results from an imbalance of oxygen supply and oxygen demand. The heart is an aerobic organ whose capacity for anaerobic glycolysis is limited; it makes use of oxygen avidly and efficiently, extracting 70% to 80% of the oxygen from coronary arterial blood.1 Because the heart extracts oxygen nearly maximally independent of demand, any increases in demand must be met by commensurate increases in coronary blood flow.
Classically, myocardial ischemia has been divided into categories including stable angina, unstable angina, and myocardial infarction. Typical angina is exertional, and is relieved promptly by rest or nitroglycerin. Stable angina occurs reproducibly with a similar level of exertion, in a pattern that has not changed over the past 6 months. Acute coronary syndromes comprise unstable angina and myocardial infarction. Unstable angina consists of ischemic symptoms which are more frequent, severe, or prolonged than the patient’s usual angina, are more difficult to control with drugs, or are occurring at rest or with minimal exertion. Cardiac biomarkers are not elevated. Myocardial infarction has been classified as “transmural” and “nontransmural,” but this division has been largely abandoned due to the recognition that electrocardiographic criteria are neither sensitive nor specific to make this distinction.
Acute coronary syndromes were previously classified into Q-wave myocardial infarction, non-Q-wave myocardial infarction, and unstable angina. More recently, classification has shifted and has become based on the initial electrocardiogram: patients are divided into three groups: those with ST elevation (STEMI), without ST elevation but with enzyme evidence of myocardial damage (non-ST elevation MI, or non-ST elevation myocardial infarction [NSTEMI]), and those with unstable angina. Classification according to presenting electrocardiogram coincides with current treatment strategies, since patients presenting with ST elevation benefit from immediate reperfusion and should be treated with fibrinolytic therapy or urgent revascularization, whereas fibrinolytic agents are not effective in other patients with acute coronary syndromes. The discussion of myocardial infarction in this chapter follows this schematization.
Myocardial ischemia results from an imbalance between oxygen supply and demand. The myocardial requirement for oxygen, and hence for oxygenated blood, is affected by three major variables: heart rate, myocardial wall stress, and contractility. Myocardial wall stress is a function of the radius, and the intraventricular pressure, which is highly dependent on ventricular afterload (see Fig. 37-1).
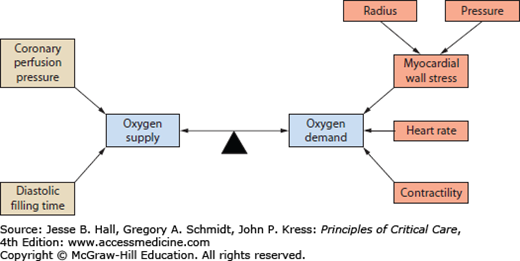
Coronary blood flow depends on coronary perfusion pressure and filling time. Since coronary perfusion occurs primarily in diastole, the relevant pressure gradient is aortic diastolic pressure minus left ventricular diastolic pressure. Filling time is directly related to heart rate.
Myocardial ischemia usually develops in the setting of obstructive atherosclerotic coronary artery disease, which limits blood supply. The pathophysiology of unstable coronary syndromes and myocardial infarction (MI) usually involves dynamic partial or complete occlusion of an epicardial coronary artery because of acute intracoronary thrombus formation.2
A number of factors in critically ill patients could increase myocardial oxygen demand, including tachycardia, hypertension, and increased catecholamines. Similarly, many factors could contribute to limitation of oxygen supply, particularly in the setting of hemodynamic instability. These factors include hypotension, decreasing coronary perfusion pressure, and tachycardia, limiting diastolic filling time. In addition, anemia and hypoxemia can limit the amount of oxygen delivered to the heart. Coronary vasospasm may also play a role in some patients. Elevation of left ventricular pressures by heart failure can both increase demand and reduce coronary perfusion pressure.
Thus, critically ill patients, usually those with at least some component of obstructive coronary artery disease, may develop myocardial ischemia on a hemodynamic basis, with variable contributions of increased demand and decreased supply. On the other hand, catecholamine surges, hemodynamic changes, and inflammatory processes may predispose to rupture of preexisting atherosclerotic plaques. Making the distinction is vital because the treatment is completely different. In the former case, treatment is aimed at decreasing the oxygen requirement of the myocardium by eliminating provocative stimuli and controlling heart rate and blood pressure, and on optimizing oxygenation and hemoglobin concentration. Relief of myocardial ischemia by these measures usually results in prompt restoration of left ventricular function without significant cellular damage, since the obstruction to flow is ordinarily fixed and not total. If plaque rupture is playing a role, then simply removing or lessening stimuli that increase myocardial oxygen requirements may not be sufficient to increase the myocardial oxygen supply:demand ratio, and unless attempts are made to reestablish coronary blood flow, significant myocardial damage may ensue. Antithrombotic and anticoagulant strategies should be instituted, and consideration of coronary revascularization may be indicated.
RECOGNITION OF MYOCARDIAL ISCHEMIA
Myocardial ischemia is most commonly manifested as constant substernal chest tightness or pressure. The pain is typically left-sided, may radiate to the throat and jaw or to the left shoulder and left arm, and is often accompanied by acute onset of dyspnea and diaphoresis. Angina may occasionally be right-sided, interscapular, or perceived in the epigastrium.
Because other syndromes may mimic angina, it is important to consider them in the differential diagnosis. These include dissecting aortic aneurysm, pericarditis, pleuritis, pulmonary processes such as pulmonary embolism, pneumonia, and pneumothorax, gastrointestinal processes such as esophageal or peptic ulcer disease and cholecystitis, musculoskeletal pain, and costochondritis. Other heart diseases (valvular heart disease, cardiomyopathies, myocarditis), not attributable to coronary artery stenosis, may also cause substernal chest tightness and should also be included in the differential diagnosis. The presentation of ischemia in postsurgical patients may be subtle. After-effects of surgery and medication can mimic or mask the classic features of myocardial infarction such as substernal chest pain radiating to the arm, neck or jaw, dyspnea, nausea, and diaphoresis. The vigilant clinician must therefore maintain a high index of suspicion and have a low threshold for obtaining a 12-lead ECG.
The physical examination, although sometimes insensitive and nonspecific, especially in the patient with multisystem illness or with preexisting left ventricular dysfunction, may be helpful in confirming the diagnosis. Elevated jugular veins signal right ventricular diastolic pressure elevation, and the appearance of pulmonary crackles (in the absence of pulmonary disease) indicates elevated left ventricular filling pressures secondary to depressed left ventricular function. A systolic bulge occasionally can be palpated on the precordium near the apex of the heart, representing contact of an ischemic dyskinetic segment of the left ventricle with the chest wall. During the ischemic episode, auscultation may reveal the presence of a fourth heart sound, indicative of a noncompliant left ventricle. With extensive myocardial dysfunction, a third heart sound may be present. A murmur of mitral regurgitation attributable to papillary muscle dysfunction may also emerge.
The electrocardiographic (ECG) abnormalities in myocardial ischemia vary widely and depend in large part on the extent and nature of coronary stenosis and the presence of collateral blood flow to ischemic zones. With acute total occlusion of a coronary artery, the first demonstrable ECG changes are peaked T waves changes in the leads reflecting the anatomic area of myocardium in jeopardy. As total occlusion continues, there is elevation of the ST segments in the same leads. With continued occlusion, there is an evolution of ECG abnormalities, with biphasic and then inverted T waves. If enough myocardium is infarcted, Q waves, which represent unopposed initial depolarization forces away from the mass of infarcted myocardium, which has lost electrical activity and no longer contributes to the mean QRS voltage vector may appear. The formation of Q waves is accompanied by a decrease in the magnitude of the R waves in the same leads, representing diminution of voltage in the mass of infarcted myocardium. Indeed, loss of R wave voltage, revealed by comparison with previous ECG tracings, may be the only ECG evidence for the presence of permanent myocardial damage. It is important to note that QRS voltage can be affected by multiple factors, such as lead placement, body position, QRS axis shifts, and pericardial and thoracic abnormalities that may shield the electrical activity of the heart. These conditions are frequently encountered in patients in the ICU and should be taken into consideration in interpretation of Q waves and R waves.
Extension of an inferior MI to the posterior segment can be detected by enhancement of R waves in the anterior chest leads, since these forces are now less opposed by posterior forces. True posterior infarction can be subtle, since the only signs may be prominent R waves, tall upright T waves and depressed ST segments in leads V1 and V2. Involvement of the right ventricle in inferior MI is also not readily detected on the standard 12-lead ECG because of the small mass of the right ventricle relative to the left ventricle and because of the positioning of the standard precordial leads away from the right ventricle. RV infarction may be detected by ST elevation in recordings from right precordial leads, particularly V4R.3
Subtotal occlusion of an epicardial coronary artery may not result in ST elevation, but rather in ST depression or only T wave changes in the leads reflecting the involved myocardium. These findings are less specific for myocardial ischemia than ST elevation, as they may also be caused by a myriad of factors besides ischemia, including cardioactive drugs, in particular digoxin, and electrolyte disorders, in particular hypokalemia. Left ventricular hypertrophy and acute left ventricular pressure overload, as might occur in hypertensive crisis, may also result in ST depression—the so-called strain pattern. Supraventricular tachycardias have also been shown to result in ST depression, even in the absence of coronary artery disease. In the presence of preexisting T-wave abnormalities, ST segment, or T wave changes are even less specific for ischemia. Ischemia may also be indicated by previously flattened or inverted T waves that revert to upright—the so-called pseudonormalization of T waves.
The clinician must also be careful not to be fooled by electrocardiographic “imposters” of acute infarction, which include pericarditis, J-point elevation, Wolff-Parkinson-White syndrome, and hypertrophic cardiomyopathy. In pericarditis, ST segments may be elevated, but the elevation is diffuse and the morphology of the ST segments in pericarditis tends to be concave upward, while that of ischemia is convex. Pericarditis may also be distinguished from infarction by the presence of PR segment depression in the inferior leads (and also by PR segment elevation in lead aVR).4
Recent interest has focused on “silent” myocardial ischemia, that is, objective ECG evidence of myocardial ischemia that is not associated with angina or with anginal equivalents.5 Silent myocardial ischemia may be an incidental observation on a cardiac monitor or on a routine ECG, and consists of transient ST segment depression that may last several minutes or even hours. The frequency of episodes of ST segment depression correlates with the severity of coronary artery disease in patients with known coronary artery disease or a history of angina.
Decreased left ventricular function has been associated with episodes of silent ST depression.6,7 In patients monitored with pulmonary artery (PA) catheters, silent ischemia may be manifested by increased pulmonary artery occlusion pressures, reflecting increased left ventricular end-diastolic pressure (LVEDP). Echocardiography may demonstrate transient wall motion abnormalities and diminished diastolic compliance. These signs of left ventricular dysfunction may precede ST segment changes.6,7
It is important to note that not all episodes of transient ST segment depression are attributable to silent ischemia. Nevertheless, should this finding be observed on the cardiac monitor, especially in association with transient elevation of left ventricular filling pressures, it is prudent to consider the possibility of myocardial ischemia as a potential factor complicating the course of the critically ill patient, and to consider additional diagnostic measures as above.
Measurement of enzymes released into the serum from necrotic myocardial cells after infarction can aid in the diagnosis of myocardial infarction.8 The classic biochemical marker of acute myocardial infarction is elevation of creatine phosphokinase (CPK) levels. The CPK MB isoenzyme is found primarily in cardiac muscle, and only small amounts are present in skeletal muscle and brain. CK released from the myocardium begins to appear in the plasma 4 to 8 hours after onset of infarction, peaks at 12 to 24 hours, and returns to baseline at 2 to 4 days. The magnitude of the increase in serum CK level and the rate at which it rises and falls are a function of the total mass of myocardium affected, the extent and nature of coronary occlusion (eg, total or subtotal occlusion), the rate of washout from the infarcted myocardium, and the clearance from the body. To be diagnostic for MI, the total plasma CK value must exceed the upper limit of normal, and the fraction consisting of the MB isoenzyme must exceed a certain value (usually >5%, but depends on the CK-MB assay used).
A newer serologic test for the detection of myocardial damage employs measurement of cardiac troponins.8 Troponin T and troponin I are constituents of the contractile protein apparatus of cardiac muscle, and are more specific than the conventional CPK-MB assays for the detection of myocardial damage. Their use is becoming more widespread, and has superseded the use of CPK MB in most settings.8 Troponins are also more sensitive for the detection of myocardial damage, and troponin elevation in patients without ST elevation (or in fact, without elevation of CPK-MB) identifies a subpopulation at increased risk for complications. Rapid point-of-care troponin assays, which have become available in the past few years, have further extended the clinical utility of this marker. Troponins may not be elevated until 6 hours after an acute event, and so critical therapeutic interventions should not be delayed pending assay results. Once elevated, troponin levels can remain high for days to weeks, limiting their utility to detect late reinfarction.
One challenge with use of troponins in the intensive care unit is that their elevation may not be confined to acute coronary syndromes. A number of other conditions prevalent in the critical care setting, including sepsis, burns, pulmonary embolism, myocarditis, and renal failure, have been associated with increases in troponin, albeit at levels lower than those usually seen with large myocardial infarctions.9 Detectable troponin levels in critically ill patients, although they usually emanate from myocardial cells, may not always represent either irreversible cell death or myocardial ischemia. Endotoxin, cytokines, and other inflammatory mediators, along with catecholamines and conditions such as hypotension, inotropes or hypoxia may cause the breakdown of cytoplasmic troponin into smaller fragments that can pass through endothelial monolayers and subsequently be detected by sensitive assays for troponin.10 In any event, isolated troponin elevation in the absence of ECG changes or other clinical signs of ischemia should be evaluated in the clinical context. In some settings, echocardiography to evaluate for new wall motion abnormalities may be useful.
To the physician confronted with a critically ill patient, echocardiography can be a key element in successful differential diagnosis.11 Echocardiography is simple, safe, and permits systemic interrogation of cardiac chamber size, left and right ventricular function, valvular structure and motion, atrial size, and the anatomy of the pericardial space. The presence of segmental left ventricular wall motion abnormalities suggests compromise of blood flow to those segments.12 Doppler interrogation can be used for noninvasive assessment of right and left ventricular filling pressures, pulmonary artery pressures, stroke volume, and cardiac output.
Echocardiography is particularly useful in the evaluation of patients with acute heart failure or suspected cardiogenic shock, and early echocardiography should be routine.13 Expeditious evaluation of global and regional left ventricular performance is crucial for management of congestive heart failure, with or without suspected myocardial ischemia.
Echocardiography is also extremely valuable for the rapid diagnosis of mechanical causes of shock after myocardial infarction such as papillary muscle rupture and acute mitral regurgitation, acute ventricular septal defect, and free wall rupture and tamponade.14 In some cases, echocardiography may reveal findings compatible with right ventricular infarction. Echocardiography can also reveal alternative diagnoses, such as valvular abnormalities, pericardial tamponade, or hypertrophic cardiomyopathy. Acute right heart failure, manifested by a dilated and hypokinetic right ventricle without hypertrophy suggestive of chronic pulmonary hypertension, can suggest pulmonary embolism.15
Transthoracic echocardiographic images may be suboptimal due to a poor acoustic window in critically ill patients, particularly those who are obese, have chronic lung disease, or are on positive pressure ventilation. Contrast echocardiography may be used to improve image quality.16 Transesophageal echocardiography (TEE) can also provide better visualization, particularly of valvular structures, and can be performed safely at the bedside.
In patients with hemodynamic instability that does not improve relatively quickly with simple therapeutic maneuvers, invasive hemodynamic monitoring should be considered. Pulmonary artery catheterization (PAC) provides simultaneous assessment of filling pressures and cardiac output, and can be quite useful for differential diagnosis in critically ill patients. In patients with hypoxemia and pulmonary infiltrates on chest x-ray, a frequent dilemma in ICU patients, PAC may be used to differentiate cardiac from pulmonary causes. Right heart catheterization is also quite useful in the differential diagnosis of shock. Hemodynamic profiles of patients with different forms of shock are shown in Table 37-1. It is important to recognize the possibility of mixed forms of shock in critically ill patient. For example, patients with myocardial infarction, even in the presence of significant left ventricular dysfunction and suspected cardiogenic shock, can be relatively volume depleted, perhaps due to diaphoresis and/or vomiting.13,17
Use of Right Heart Catheterization to Diagnose the Etiology of Shock
| Diagnosis | Pulmonary Artery Occlusion Pressure | Cardiac Output | SVR | Miscellaneous Comments |
|---|---|---|---|---|
| Cardiogenic Shock | ||||
| Cardiogenic shock due to myocardial dysfunction | ⇑⇑ | ⇓⇓ | ⇑⇑ | Usually extensive infarction (>40% of LV), severe cardiomyopathy, or myocarditis |
| Cardiogenic shock due to mechanical defects | ||||
| Acute ventricular septal defect | ⇑ | ⇓⇓ | ⇑⇑ | Oxygen “step-up” at RV level |
| Acute mitral regurgitation | ⇑⇑ | Forward CO ⇓⇓ | ⇑ | V waves in PAOP tracing |
| Right ventricular infarction | Normal or ⇓ | ⇓⇓ | ⇑⇑ | Elevated RA and RV filling pressures with low or normal PAOP |
| Extracardiac Obstructive Forms of Shock | ||||
| Pericardial tamponade | ⇑ | ⇓ or ⇓⇓ | ⇑⇑ | RA mean, RV end-diastolic pulmonary capillary wedge mean pressures are elevated and within 5 mm Hg of one another |
| Massive pulmonary embolism | normal or ⇓ | ⇓⇓ | ⇑⇑ | Usual finding is elevated right-sided pressures |
| Hypovolemic Shock | ⇓⇓ | ⇓⇓ | ⇑⇑ | |
| Distributive Forms of Shock | ||||
| Septic shock | ⇓ or normal | ⇑ or normal, rarely ⇓ | ⇓⇓ | |
| Anaphylactic shock | ⇓ or normal | ⇑ or normal | ⇓⇓ | |
Hemodynamic monitoring can also be useful in the diagnosis of mechanical complications of infarction, although most causes are more easily identified with echocardiography. Right heart catheterization may reveal a step-up in hemoglobin oxygen saturation diagnostic of ventricular septal rupture. The waveform of the PAOP tracing may reveal a prominent V wave (10 mm Hg above the mean PAOP is regarded as significant) suggesting severe mitral regurgitation, although V waves may be present in acute ventricular septal rupture as well. Equalization of diastolic filling pressures may suggest pericardial tamponade. The hemodynamic profile of RV infarction includes high right-sided filling pressures in the presence of normal or low occlusion pressures.18
Right heart catheterization is most useful, however, to optimize therapy in unstable patients. Infusions of vasoactive agents need to be titrated carefully in patients with myocardial ischemia to maximize coronary perfusion pressure with the least possible increase in myocardial oxygen demand. Invasive hemodynamic monitoring can be extremely useful in allowing optimization of therapy in these unstable patients, because clinical estimates of filling pressure can be unreliable19; in addition, changes in myocardial performance and compliance and therapeutic interventions can change cardiac output and filling pressures precipitously. Optimization of filling pressures and serial measurements of cardiac output (and other parameters, such as mixed venous oxygen saturation) allow for titration of the dosage of inotropic agents and vasopressors to the minimum dosage required to achieve the chosen therapeutic goals. This minimizes the increases in myocardial oxygen demand and arrhythmogenic potential.20 Although PAC is useful to obtain indices of cardiac output to guide the use of inotropic agents and to obtain filling pressures on a serial basis to guide the use of both vasopressors and vasodilators, other methods of obtaining these indices are reasonable as well.
MANAGEMENT OF ANGINA
As previously noted, myocardial ischemia results from an imbalance of myocardial oxygen supply and demand. Patients with a history of stable angina who develop chest pain while in the critical care setting are best treated by removal of provocative stimuli that increase myocardial oxygen consumption or lead to compromised coronary blood flow, if these factors can be identified. For example, correction of hypoxia, anemia, hypovolemia, tachycardia, or labile hypertension may be sufficient to control anginal episodes. Often overlooked are fever, infection, anxiety, stress, activity, and the work of breathing. Antianginal medications the patient was receiving before hospitalization should be continued, and the doses possibly increased.
In instances of refractory angina or where provocative stimuli cannot be ameliorated, it may be necessary to perform coronary angiography and revascularization of the culprit vessels (preferably percutaneously), especially if the myocardial ischemia is complicating patient management.
Aspirin is the best known and the most widely used of all the antiplatelet agents because of its low cost and relatively low toxicity. Use of salicylates to treat coronary artery disease in the United States was first reported in 1953.21 Aspirin inhibits the production of thromboxane A2 by irreversibly acetylating the serine residue of the enzyme prostaglandin H2 synthetase.
Aspirin has also been shown to be beneficial in preventing cardiovascular events when administered as secondary prevention in patients after acute myocardial infarction and as primary prevention in subjects with no prior history of vascular disease.22 Doses of aspirin used in cardiovascular disease range between 81 mg and 325 mg daily.23 Despite the fact aspirin blocks thromboxane preferentially to prostacyclin at low doses and thus has a more profound antiplatelet effect, high-dose aspirin has been found to be as effective as low-dose aspirin in cardiovascular prevention, which may suggest that besides its antiplatelet effects, anti-inflammatory effects of aspirin play a role as well.24 Once begun, aspirin should probably be continued indefinitely. Toxicity with aspirin is mostly gastrointestinal; enteric-coated preparations may minimize these side effects.
Nitroglycerin is a mainstay of therapy for angina because of its efficacy and rapid onset of action. The most important antianginal effect of nitroglycerin is preferential dilation of venous capacitance vessels, decreasing venous return. A reduction in myocardial oxygen demand and consumption results from the reduction of LV volume and arterial pressure primarily due to reduced preload.25 At higher doses, in some patients, nitroglycerin relaxes arterial smooth muscle as well, causing a modest decrease in afterload, which also contributes to wall stress.25 In addition, nitroglycerin can dilate epicardial coronary arteries, and nitroglycerin redistributes coronary blood flow to ischemic regions by dilating collateral vessels. Nitroglycerin has antithrombotic and antiplatelet effects as well.
The quickest route of administration of nitroglycerin is sublingual. Sublingual doses of 0.4 mg may be administered every 5 to 10 minutes to a total of three doses, if required to control pain. Topical or oral nitrates may be used for chronic therapy.
In patients with unstable angina, if sublingual nitroglycerin does not cause chest pain to resolve completely, intravenous nitroglycerin should be administered, starting at a dose of 10 to 20 µg/min. This dose may be titrated upward as tolerated in increments of 10 to 20 µg/min every 5 to 10 minutes. An upper limit of 400 µg/min is usually accepted as maximal; above this dose there is usually no further clinical response. Because of its hemodynamic actions, systemic blood pressure may fall after nitroglycerin administration, so frequent blood pressure checks are required; untoward decreases in blood pressure can compromise coronary perfusion. Hypotension usually resolves with Trendelenburg position and/or intravenous saline boluses.
The rationale for administration of β-blockers during ischemic episodes derives from their negative chronotropic and negative inotropic properties. Heart rate and contractility are two of the three major determinants of myocardial oxygen consumption. By altering these variables, myocardial ischemia can be attenuated significantly.26 These agents are particularly effective in patients with angina who remain tachycardic or hypertensive (or both) and in patients with supraventricular tachycardia complicating myocardial ischemia. Rapid control can be achieved by intravenous administration of metoprolol, a β1-selective blocker, in 5 mg increments every 5 minutes up to 15 mg. Thereafter, 25 to 50 mg every 6 hours can be given orally.
β-Blockers should be used with caution in patients with marginal blood pressure, preexisting bradycardia, AV nodal conduction disturbances, and evidence for left ventricular failure, as well as those with bronchospastic disease. A short-acting intravenous β-blocker, such as esmolol, may be the preferred agent in patients who have the potential for hemodynamic instability or who have relative contraindications.
Non-dihydropyridine calcium channel blockers (verapamil and diltiazem) also have negative chronotropic and inotropic effects, and can be used to control myocardial oxygen demand in patients with ischemia. Both can be given as intravenous boluses, starting with low doses (diltiazem 10-20 mg, verapamil 2.5 mg), and can then be infused continuously.
Calcium channel blockers are particularly useful in the setting of coronary vasospasm, because they cause direct dilation of coronary vascular smooth muscle. Vasospasm can produce variant angina in patients with mild or no coronary artery disease (Prinzmetal’s angina), or aggravate ischemia in patients with atherosclerotic coronary stenoses that are subcritical but serve as sites of vasospasm, possibly as a consequence of abnormalities of the underlying smooth muscle or derangements in endothelial physiology.27 The illicit use of cocaine is increasingly being recognized as a cause of coronary vasospasm leading to angina and myocardial ischemia. Coronary vasospasm usually presents with ST elevation associated with chest pain, and can be difficult to differentiate from vessel closure due to coronary thrombosis. Consideration of the clinical setting, rapid fluctuation of ST segments, and prompt resolution with nitrates can provide useful clues. Variant angina attributable to vasospasm responds well to treatment with calcium channel blockers.
Angiotensin converting enzyme (ACE) generates angiotensin II from angiotensin I and also catalyzes the breakdown of bradykinin. Thus ACE inhibitors can decrease circulating angiotensin II levels and increase levels of bradykinin, which in turn stimulates production of nitric oxide by endothelial nitric oxide synthase. In the vasculature, ACE inhibition promotes vasodilation, and tends to inhibit smooth muscle proliferation, platelet aggregation, and thrombosis.
The major hemodynamic effect of ACE inhibition is afterload reduction, which is most important as an influence of myocardial oxygen demand in patients with impaired left ventricular function. A recent study, however, has demonstrated that ACE inhibition may be beneficial to prevent recurrent events in high-risk patients. The HOPE trial of 9297 patients with documented vascular disease or atherosclerosis risk factor showed that ramipril (target dose 10 mg/d) reduced cardiovascular death, myocardial infarction (MI), and stroke by 22% compared to placebo.28 Patients were normotensive at the start of the trial, and the magnitude of benefit observed was not explained by the modest reduction in blood pressure (2-3 mm Hg).28 The ACC/AHA guidelines recommend use of ACE inhibitors in most cases as routine secondary prevention for patients with known CAD, particularly in diabetics without severe renal disease.29
Extensive epidemiologic, laboratory, and clinical evidence provides a convincing relationship between cholesterol and coronary artery disease. Total cholesterol level has been linked to the development of CAD events with a continuous and graded relation, with a close association with LDL cholesterol.30 Numerous large primary and secondary prevention trials have shown that LDL cholesterol lowering is associated with a reduced risk of coronary disease events. The earliest lipid-lowering trials used bile-acid sequestrants (cholestyramine), fibric acid derivatives (gemfibrozil and clofibrate), or niacin in addition to diet, achieving a reduction in total cholesterol of 6% to 15%, accompanied by a consistent trend toward a reduction in fatal and nonfatal coronary events.31
HMG-CoA reductase inhibitors (statins) produce larger reductions in cholesterol, with more impressive clinical results. Statins have been demonstrated to decrease the rate of adverse ischemic events and mortality when used both as primary prevention in high-risk patients,32,33 and as secondary prevention in patients with documented CAD.34-36 The goal of treatment is an LDL cholesterol level less than 100 mg/dL,37 although there appears to be a linear relationship between LDL levels and events, and many clinicians recommend an LDL goal of <70 mg/dL, especially for secondary prevention. Maximum benefit may require management of other lipid abnormalities (elevated triglycerides, low HDL cholesterol) and treatment of other atherogenic risk factors.
Intra-Aortic Balloon Pump Counterpulsation: When angina remains refractory to maximal medical therapy, intra-aortic balloon pump counterpulsation may be considered. The intra-aortic balloon pump (IABP) is a device that is inserted via the femoral artery into the descending thoracic aorta just distal to the aortic arch. A 40-mL balloon at the tip of the catheter is inflated in diastole by a pneumatic pump in synchrony with closure of the aortic valve, and is deflated on opening of the aortic valve. Inflation and deflation are gated to the R and T waves on the ECG or to the arterial pressure recording. By deflating during ventricular systole, ventricular afterload is reduced, resulting in significant decreases in myocardial wall stress and significant decreases in myocardial oxygen requirements.38 Furthermore, inflation during diastole augments coronary blood flow by increasing coronary perfusion pressure. The main way in which an IABP relieves myocardial ischemia is by decreasing oxygen demand through afterload reduction.39
Use of an IABP is indicated in unstable angina when the angina and attendant ECG abnormalities are persistent and refractory to maximal pharmacologic therapy. An IABP also may be inserted in patients who are stable and have undergone angiography but in whom precarious coronary lesions (eg, left main coronary artery stenosis) have been identified. Typically, these patients are maintained on the device while awaiting surgery or angioplasty.
Although insertion of an IABP can result in immediate and dramatic relief of myocardial ischemia, there are potential complications,38 including aortic dissection, bleeding, femoral neuropathies, renal failure from renal artery occlusion, arterial thrombi and emboli, limb ischemia, and line sepsis. These potential complications must be weighed in determining whether an IABP should be inserted.
Coronary Angiography: If anginal symptoms persist despite maximal medical therapy, coronary angiography with an aim toward possible revascularization should be considered. Frequent anginal episodes or episodes that are difficult to control with conventional antianginal medications may suggest impending infarction. Under these circumstances, early angiography is indicated. In cases in which the patient is stabilized readily with pharmacologic agents, angiography may be delayed or even deferred altogether. One must keep in mind that coronary angiography is not a therapeutic intervention, but a diagnostic test. Angiography is of little tangible value if there are no viable revascularization options. The optimal timing of angiography in patients with non-ST elevation acute syndromes is a separate and evolving issue that will be considered in the section on NSTEMI.
ST ELEVATION MYOCARDIAL INFARCTION
Symptoms suggestive of MI may be similar to those of ordinary angina but are usually greater in intensity and duration. Nausea, vomiting, and diaphoresis may be prominent features, and stupor and malaise attributable to low cardiac output may occur. Compromised left ventricular function may result in pulmonary edema with development of pulmonary bibasilar crackles and jugular venous distention; a fourth heart sound can be present with small infarcts or even mild ischemia, but a third heart sound is usually indicative of more extensive damage.
Patients presenting with suspected myocardial ischemia should undergo a rapid evaluation. A 12-lead electrocardiogram should be performed and interpreted expeditiously. Initial therapy should include aspirin, 160 to 325 mg orally, sublingual nitroglycerin (unless systolic pressure is <90 mm Hg), and usually oxygen, even though hard evidence for benefits of oxygen in patients without hypoxia is not compelling.40,41 Opiates should be used to relieve pain, and also to reduce anxiety, the salutary effects of which have been known for decades and must not be underestimated. It is also important to provide reassurance to the patient.
ST-segment elevation of at least 1 mV in two or more contiguous leads provides strong evidence of thrombotic coronary occlusion, the patient should be considered for immediate reperfusion therapy. The diagnosis of STEMI can be limited in the presence of preexisting left bundle-branch block (LBBB) or a permanent pacemaker. Nonetheless, new LBBB with a compatible clinical presentation should be treated as acute myocardial infarction and treated accordingly. Indeed, data suggest that patients with STEMI and new LBBB may stand to gain even greater benefit from reperfusion strategies than those with ST elevation.42
Early reperfusion of an occluded coronary artery is indicated for all eligible candidates. Overwhelming evidence from multiple clinical trials demonstrates the ability of thrombolytic agents administered early in the course of an acute MI to reduce infarct size, preserve left ventricular function, and reduce short-term and long-term mortality.43,44 Patients treated early derive the most benefit. Indications and contraindications for thrombolytic therapy are listed in Table 37-2. Because of the small, but nonetheless significant, risk of a bleeding complication, most notably intracranial hemorrhage, selection of patients with acute MI for administration of a thrombolytic agent should be undertaken with prudence and caution. That is of special importance in ICU patients, who may have a predisposition to bleeding complications because of multiple factors. In the surgical patient, fibrinolysis may pose a prohibitive risk and emergent coronary angiography (with percutaneous coronary intervention [PCI] as clinically indicated) may be preferable.
Indications for and Contraindications to Thrombolytic Therapy in Acute Myocardial Infarction
| Indications |
|
| Contraindications |
| Absolute |
|
| Relative |
|
In contrast to the treatment of STEMI, fibrinolytics have shown no benefit and an increased risk of adverse events when used for the treatment of unstable angina/NSTEMI.45 Based on these findings, there is currently no role for thrombolytic agents in these latter syndromes.
Thrombolytic Agents: Streptokinase was the original lytic agent used in MI, but has now been superseded by tissue plasminogen activator (t-PA), a recombinant protein that is more fibrin-selective than streptokinase and produces a higher early coronary patency rate (70%-80%). The Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries (GUSTO) trial compared SK to t-PA in 41,101 patients with STEMI, and demonstrated a small but significant survival benefit for t-PA (1.1% absolute, 15% relative reduction).46 The GUSTO angiographic substudy showed that the difference in clinical efficacy resulted from the difference in patency rates.47 t-PA is usually given in an accelerated regimen consisting of a 15-mg bolus, 0.75 mg/kg (up to 50 mg) IV over the initial 30 minutes, and 0.5 mg/kg (up to 35 mg) over the next 60 minutes.
Reteplase (r-PA), is a deletion mutant of t-PA with an extended half-life, and is given as two 10-mg boluses 30 minutes apart. Reteplase was originally evaluated in angiographic trials which demonstrated improved coronary flow at 90 minutes compared to t-PA, but subsequent trials showed similar 30-day mortality and bleeding rates.48
Tenecteplase (TNK-t-PA) is a genetically engineered t-PA mutant with amino acid substitutions that result in prolonged half-life, resistance to plasminogen-activator inhibitor-1, and increased fibrin specificity. TNK-t-PA is given as a single bolus, adjusted for weight. A single bolus of TNK-t-PA has been shown to produce coronary flow rates identical to those seen with accelerated t-PA, with equivalent 30-day mortality and bleeding rates.49
Because these newer agents in general have equivalent efficacy and side effect profiles, at no current additional cost compared to t-PA, and because they are simpler to administer, they have gained popularity. An ideal fibrinolytic agent would have greater fibrin specificity, slower clearance from the circulation, and more resistance to plasma protease inhibitors, but has not yet been developed.
The major advantages of primary PCI over thrombolytic therapy include a higher rate of normal (TIMI grade 3)44 flow, lower risk of intracranial hemorrhage and the ability to stratify risk based on the severity and distribution of coronary artery disease. Patients ineligible for fibrinolytic therapy obviously should be considered for primary PCI. In addition, data from several randomized trials have suggested that PCI is preferable to thrombolytic therapy for AMI patients at higher risk, including those over 75 years old, those with anterior infarctions, and those with hemodynamic instability.50,51 The largest of these trials is the GUSTO-IIb Angioplasty Substudy, which randomized 1138 patients. At 30 days, there was a clinical benefit in the combined primary end points of death, nonfatal reinfarction, and nonfatal disabling stroke in the patients treated with PTCA compared to t-PA, but no difference in the “hard” end points of death and myocardial infarction at 30 days.51
Meta-analyses comparing direct PCI with thrombolytic therapy found lower rates of mortality and reinfarction among those receiving direct PCI.52,53 Thus, direct angioplasty, if performed in a timely manner (ideally within 60 minutes) by highly experienced personnel, may be the preferred method of revascularization since it offers more complete revascularization with improved restoration of normal coronary blood flow and detailed information about coronary anatomy. There are certain subpopulations in which primary PCI is clearly preferred, and other populations in which the data are suggestive of benefit. These subsets are listed in Table 37-3. More important than the method of revascularization is the time to revascularization, and that this should be achieved in the most efficient and expeditious manner possible.54 It is important to keep in mind that early, complete, and sustained reperfusion after myocardial infarction is known to decrease 30-day mortality. The preferred method for reperfusion in STEMI is PCI only if it can be done within a timely manner. Practical considerations regarding transport to a PCI capable facility should be carefully reviewed before foregoing fibrinolytics for PCI. Early recognition and diagnosis of STEMI are key to achieving the desired door-to-needle (or medical contact–to-needle) time for initiation of fibrinolytic therapy of 30 minutes or door-to-balloon (or medical contact–to-balloon) time for PCI under 90 minutes.41 Achieving reperfusion in timely matter correlates with improvement in ultimate infarct size, left ventricular function, and survival.55,56 The ultimate goal is to restore adequate blood flow through the infarct-related artery to the infarct zone as well as to limit microvascular damage and reperfusion injury. The latter is accomplished with adjunctive and ancillary treatments that will be discussed below.
Situations in Which Primary Angioplasty Is Preferred in Acute Myocardial Infarction
| Situations in Which PTCA Is Clearly Preferable to Thrombolytics |
|
| Situations in Which PTCA May Be Preferable to Thrombolytics |
|
Coronary Stenting: Primary angioplasty for acute myocardial infarction results in a significant reduction in mortality but is limited by the possibility of abrupt vessel closure, recurrent in-hospital ischemia, reocclusion of the infarct related artery, and restenosis. The use of coronary stents has been shown to reduce restenosis and adverse cardiac outcomes in both routine and high-risk PCI.57 The PAMI Stent Trial was designed to test the hypothesis that routine implantation of an intracoronary stent in the setting of myocardial infarction would reduce angiographic restenosis and improve clinical outcomes compared to primary balloon angioplasty alone. This large, randomized, multicenter trial involving 900 patients did not show a difference in mortality at 6 months but did show improvement in ischemia-driven target vessel revascularization and less angina in the stented patients compared to balloon angioplasty alone.58 Despite the lack of definite data demonstrating mortality benefit, virtually all of the trials investigating adjunctive therapy for STEMI have employed a strategy of primary stenting, and stenting is becoming the default strategy. Whether to use a bare metal stent or a drug eluting stent in acute MI is a question that has not yet been addressed definitively by clinical trials; selection is currently based on both patient and angiographic characteristics.
Aspirin: Aspirin is the best known and the most widely used of all the antiplatelet agents because of low cost and relatively low toxicity. Aspirin has been shown to reduce mortality in acute infarction to the same degree as fibrinolytic therapy, and its effects are additive to fibrinolytics.59 In addition, aspirin reduces the risk of reinfarction. Unless contraindicated, all patients with a suspected acute coronary syndrome (STEMI, NSTEMI, unstable angina) should be given aspirin as soon as possible.
Related posts:






Full access? Get Clinical Tree


