KEY POINTS
Right heart syndromes (RHS) as a cause of hemodynamic instability and shock are less common than left heart dysfunction, but recognizing them requires a high level of vigilance.
RHS result from a combination of pressure or volume overload and impaired RV contractility. Progression to acute cor pulmonale (the combination of acute pulmonary hypertension with profound RV systolic and diastolic dysfunction) results in spiraling end-organ dysfunction.
Clues to recognizing RHS as a cause of shock include a history of a condition that is associated with pulmonary hypertension, elevated neck veins, peripheral edema greater than pulmonary edema, or a right-sided third heart sound, in addition to electrocardiographic, radiographic, and echocardiographic findings.
Plasma biomarkers are nonspecific but echocardiography is extremely valuable, not only for demonstrating the presence of RHS, but also for guiding hemodynamic management.
Progressive right heart shock can be worsened by excessive fluid infusion, concomitant left ventricular failure, inappropriate application of extrinsic positive end-expiratory pressure (PEEP) and hypoxia.
The drug of choice for resuscitation to reduce systemic oxygen demand while improving oxygen delivery is dobutamine, initially infused at 5 μg/kg per minute. Systemically active vasoconstrictors may provide additional benefit.
Inhaled nitric oxide or prostacyclin and oral PDE-inhibitors (eg, sildenafil) or extracorporeal mechanical assist devices may be beneficial in improving pulmonary hemodynamics and oxygenation, but may not improve survival.
In the majority of patients with shock due to “pump failure,” assessment is focused appropriately on the left ventricle. However, in a substantial minority of patients, right heart dysfunction is the cause of shock. Examples include acute pulmonary embolism (PE), other causes of acute right heart pressure overload (eg, acute respiratory distress syndrome [ARDS] treated with positive pressure ventilation), acute deterioration in patients with chronic pulmonary hypertension, and right ventricular infarction. Although right ventricular infarction differs from the other right heart syndromes (RHS) in that the pulmonary artery pressure is not high, in many other regards right ventricular infarction resembles the other syndromes, so we will consider them together. Failure to consider the right heart in the differential diagnosis of shock risks incomplete or inappropriate treatment of the shock. It would be hard to overemphasize the importance of echocardiography, both in aiding the recognition of the right heart syndromes and in guiding management. In this chapter, we review the notable features that distinguish the right heart from the left, describe the themes that unify the acute RHS and allow their recognition, discuss the pathophysiology and differential diagnosis of RHS, and review their management.
RIGHT VENTRICULAR PHYSIOLOGY
The right ventricle (RV) has long been considered the “forgotten ventricle,” because under normal pressure and volume loading conditions the RV is thought to function as a passive conduit for systemic venous return. When the pulmonary vasculature is normal, right ventricular performance has little impact on the maintenance of cardiac output. In animal models, complete ablation of the right ventricular free wall has little effect on venous pressures.
Despite the requirement for an equal, average cardiac output between the left and right ventricles, the bioenergetic requirement for RV ejection is approximately one fifth of the left ventricle (LV). This is in large part accounted for by the significant difference in downstream vascular resistance between the systemic and pulmonary circulations In comparison with the LV, the RV ejects into a low-resistance circuit (normally only one-tenth the resistance of the systemic arteries).
The pressure-volume relationship of the normal RV differs significantly from that of the LV. In contrast to the LV ejection, the RV ejects into the pulmonary outflow tract early during systole, continuing even after the maximal development of RV systolic pressure.1 This exaggerated “hang out” period (ventricular outflow between the onset of right ventricular pressure decline and pulmonary valve closure) optimizes pump efficiency and results in a triangular pressure-volume relationship compared with the square wave pump of the LV. Consequently RV wall stress is low under normal physiological conditions and RV coronary perfusion occurs in both diastole and systole, unlike the LV.2
The functional differences between RV and LV result from ontogenic, structural, cellular, and biochemical differences. The RV has a higher proportion of rapidly contractile α-myosin heavy chain filaments than the LV.3 Additionally, while both LV and RV are equally inotropically responsive to selective β1-adrenergic receptor (AR) agonism, RV and LV myocytes respond differentially to α1-adrenergic receptor stimulation.4 Selective α1-AR stimulation is negatively inotropic in RV trabeculae but positively inotropic in LV trabeculae.
PATHOPHYSIOLOGY OF RIGHT HEART SYNDROMES
Acute and acute-on-chronic right heart syndromes develop as a consequence of a combination of factors that that include impaired RV contractility, RV pressure overload or volume overload (Fig. 38-1).5
FIGURE 38-1
This figure illustrates the theory of right ventricular infarction in the right heart syndromes. A sudden rise in pulmonary artery pressure impedes right ventricular ejection. Right ventricular stroke volume falls, and end-diastolic and end-systolic volumes rise. Heart rate increases as the baroreceptors sense the fall in systemic blood pressure. These features of increased preload, afterload, and rate raise the right ventricular oxygen consumption. At the same time, the fall in aortic pressure lessens the driving gradient (roughly aortic pressure—right atrial pressure) for right coronary flow, reducing oxygen delivery to the right ventricle. If the rise in pulmonary artery pressure is sufficient, the right ventricle will fail. Vasoconstrictors have the potential to partially restore right ventricular function. Constriction of the systemic arteries raises left ventricular oxygen demands, but the normal left ventricle is operating with a margin of safety before the increased aortic pressure would be a problem. The higher aortic pressure drives more blood flow to the right ventricle without augmenting any of the components of right ventricular oxygen demand, thereby relieving ischemia and improving function.
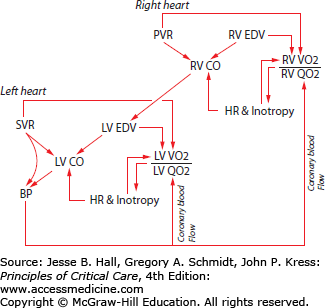
Under conditions of increased RV impedance (eg, pulmonary stenosis or pulmonary embolism) the RV pressure-volume relationship assumes a square wave appearance similar to that of the LV.6 Acute RV pressure overload leads to enhanced contractility through two mechanisms: (1) the Anrep effect (homeometric autoregulation)—an adrenergically—independent contractile enhancement and (2) the Frank-Starling mechanism. In contrast, acute volume overload evokes predominantly Starling-mediated contractile enhancement. Unlike the LV, however, even modest acute increases in RV afterload may precipitate ventricular failure. Right ventricular ejection fraction falls as Pa resistance/pressure rises and RV end-systolic and end-diastolic pressures rise. During acute Pa hypertension, RV preload, after-load, and contractile state rise at the same time that heart rate rises. These features join to raise the RV myocardial oxygen consumption. At the same time, when an acute RHS is sufficiently severe to cause systemic hypotension, coronary perfusion of the RV may fall. The combination of rising oxygen demand and falling coronary oxygen supply subjects the RV to ischemia sufficient to reduce RV contractility and reduce systolic ejection against the increased Pa pressure afterload (Fig. 38-1). The close anatomic approximation between the right and left ventricles confers a mechanical and functional interdependence in the face of right ventricular dysfunction.
These pathophysiologic derangements are different if volume and pressure loading develop more chronically. Under chronic stress conditions, RV pressure overload taxes contractile and elastic reserves more profoundly than chronic volume overload.7 Significant contractile reserve is supported by RV myocyte hypertrophy and is regulated in part by increased expression of angiotensin II, insulin-like growth factor-I, and endothelin-1.8 Ventricular hypertrophy is not uniform and is frequently associated with regional diastolic and systolic dysfunction.9 Increased cardiac output is accommodated by recruitment of previously unperfused pulmonary vessels and by distention of vessels.
LV/RV interdependence is influenced by (1) the cardiac fibroskeleton that limits acute annular distension, (2) the interventricular septum, and (3) the pericardium. As right heart volumes rise, the interventricular septum shifts progressively to the left, causing left ventricular diastolic dysfunction, further reducing systemic cardiac output and coronary perfusion pressure. Additionally, the pericardium restricts excessive acute ventricular distension while impairing diastolic filling of both the left and right heart.10 A vicious cycle ensues in which RV ischemia impairs right ventricular ejection, which without intervention leads intractably to progressive dilation of the RV and septal displacement that causes more LV diastolic dysfunction, progressive systemic hypotension, and further impairment of RV perfusion.11
This cycle has long been recognized in the acute inability of the RV to sustain a mean pulmonary artery pressure greater than about 40 mm Hg, based on studies of pulmonary hemodynamics in patients with acute PE without prior cardiopulmonary disease.1,12 There is significant evidence that even in the absence of flow limiting coronary occlusion, RV ischemia underlies acute RV failure in settings of acute pulmonary hypertension. Indirect indications include the significantly increased load tolerance of the right ventricle when aortic pressure is raised,13 and a beneficial hemodynamic response to infusion of norepinephrine.14 These findings suggest, but do not establish, that greater coronary flow driven by the higher aortic pressure enhances RV function by relieving ischemia.
Significant troponin elevation may be an early and reliable marker of right ventricular dysfunction in acute pulmonary embolism, and has been shown to predict an adverse outcome.15 Significant elevations of serum cardiac troponins T and I are thought to result from RV microinfarction.16 Histopathological evidence of myocyte necrosis and evidence of protease (calpain) activation have been described in response to acute RV pressure overload. Additionally differential gene expression patterns have been reported in rat RV myocytes after either pressure or volume overload. BNP upregulation is evident in both stressed groups. Relatively higher expression of mRNA for the inflammatory gene products of TNF-α, IL-6, pre-pro ET-1, SERCA2a, and phospholamban genes are present in volume overloaded RV myocytes.17
Clinical Clues: In the hypoperfused patient, several clinical features should suggest the possibility of an acute right heart syndrome (Table 38-1). First, any history of pulmonary hypertension raises the possibility that the new shock state represents a (potentially minor) precipitant on top of preexisting right heart compromise (acute-on-chronic pulmonary hypertension; Table 38-2). When there is no antecedent history of pulmonary hypertension, elevated neck veins, a pulsatile liver, peripheral edema out of proportion to pulmonary edema, a right-sided third heart sound, or tricuspid regurgitation should alert the intensivist that she or he may be dealing with an RHS. The pulmonic component of the second heart sound may be loud, and the time interval between the aortic (A2) and the pulmonary (P2) components of the second heart sound (A2-P2 splitting) is increased in the presence of pulmonary hypertension. However, these findings are appreciable with a binaural stethoscope in only a minority of patients with acute pulmonary embolism,18 and are probably too subjective to be useful. More sophisticated acoustic processing of digitally acquired heart sounds may provide an accurate estimation of pulmonary arterial pressures.19
Causes of Severe Pulmonary Hypertension and Acute Right Heart Syndrome
Clinical Classification of Pulmonary Hypertension
|
Despite the insensitivity of individual clinical signs to detect and diagnose acute right heart syndromes, a combination of clinical features (symptoms of deep venous thrombosis [DVT]; an alternative diagnosis is less likely than PE; heart rate >100 bpm; immobilization or surgery in the previous 4 weeks; previous DVT or PE; hemoptysis; and cancer, being treated currently or within the previous 6 months) and laboratory results, especially serum D-dimer level, can be useful in excluding pulmonary embolism as a likely cause.20 The performance characteristics of clinical prediction rules (Wells score or revised Geneva score) in combination with D-dimer assays21 for the diagnosis of PE are discussed in Chap. 39.
Electrocardiography: Electrocardiographic (ECG) evidence of RV failure in the context of pulmonary hypertension includes sinus tachycardia or atrial fibrillation, right axis deviation or a rightward shift in axis, right atrial enlargement, right ventricular hypertrophy, right bundle-branch block (RBBB), right precordial T-wave inversions (leads III and aVF or in leads V1-V4), and the S1Q3T3 pattern. Additional configurations include a Qr pattern in lead V1, S waves in lead I and aVL >1.5 mm and Q waves in leads III and aVF, but not in lead II.5 Reports of typical ECG changes vary significantly22 suggesting relative insensitivity in the performance characteristics of the 12-lead ECG in broad groups of mixed severity right heart syndrome patients. However, in patients with hemodynamically significant pulmonary embolism, the likelihood of suggestive electrocardiographic findings is probably much higher. For example, among 49 patients with PE (all of whom had RV dilation and tricuspid regurgitation by echocardiography), 37 (76%) had electrocardiographic abnormalities strongly suggestive of PE, including at least three of the following: incomplete or complete RBBB; S waves greater than 1.5 mm in leads I and aVL; shift of the precordial transition zone to V5; Q waves in leads III and aVF, but not lead II; right axis deviation or an indeterminate axis; low QRS voltage in the limb leads; or T-wave inversion in leads III and aVR or in leads V1 to V4.23 The electrocardiographic signs of right ventricular infarction are described below.
Radiography: Radiographic signs include retrosternal airspace opacification reflecting RV enlargement, prominence of the right-heart border reflecting right atrial dilation, an enlarged pulmonary artery or right ventricle, oligemia of a lobe or lung (Westermark sign), pleural or pericardial effusions and a distended azygos (or other central) vein (Figs. 38-2 and 38-3).
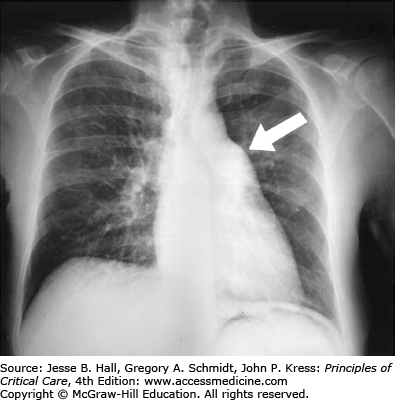
FIGURE 38-3
Chest radiograph showing a huge azygos vein (arrow) in an elderly man with shock due to acute massive pulmonary embolism. The normal vein measures less than 10 mm in transverse diameter, whereas this patient’s azygos vein measures more than 22 mm. This film also demonstrates Westermark sign (oligemia, here of all lung fields).
Contrast-enhanced computed tomography of the pulmonary vasculature (helical CT angiography) has evolved as a central diagnostic tool in the evaluation of acute right heart syndromes, particularly pulmonary thromboembolism, as discussed in Chap. 39.24-27 The sensitivities range from 53% to 89%, and specificities from 78% to 100% for single-slice helical CT diagnosis of acute PE.28 Newer multi-row detector scanners should increase sensitivity to more than 90%.29 In addition to detecting the presence of a pulmonary vascular clot, CT is able to detect RV dilation and septal shift. In a small series of patients with acute PE, CT sensitivity was 78% for detecting RV dysfunction when compared with transthoracic echocardiography (TTE).30
Cardiac magnetic resonance imaging (MRI) while time intensive, is the most accurate method for assessing RV volume and ejection fraction.31
Echocardiography: Echocardiography is of great utility in the detection of RHS and should be obtained early in the hypoperfused patient whenever one of the previously mentioned clinical indicators is present.32 Of course, most of these signs are not specific for RHS, but their recognition is important because the treatment of RHS is unique in several regards (Table 38-3). TTE is particularly useful in differentiating at the bedside right ventricular pressure overload from myocardial infarction, aortic dissection, or pericardial tamponade, all of which may be clinically indistinguishable from right ventricular pressure overload.33 Identification of a patent foramen ovale and free-floating right-heart thrombus are echocardiographic markers of particularly grave prognosis, including recurrent PE and death.33-35
Causes of RV Failure in ICU
| RV Pressure Overload, Pulmonary Hypertension, Any Cause |
| Pulmonary embolism |
| ARDS |
| Excessive PEEP, tidal volume, and alveolar pressure |
| Air, amniotic, fat, or tumor microembolism |
| Sepsis (rarely) |
| Pulmonary leukostasis, leukoagglutination |
| Extensive lung resection |
| Drugs (eg, heparin-protamine reaction) |
| Hypoxia |
| Reduced RV Contractility |
| RV infarction |
| Sepsis |
| RV cardiomyopathy |
| Myocarditis pericardial disease; LVAD; post-CPB; postcardiac surgery/transplantation |
| RV-Volume Overload |
| Tricuspid and pulmonary regurgitation; intracardiac shunts |
Furthermore, TTE may be used to estimate pulmonary artery pressures and assess right ventricular function,36 thereby allowing for rapid initiation of appropriate therapeutic interventions. Echocardiography may also provide indirect evidence of pulmonary embolism by demonstrating a specific pattern of right ventricular dysfunction characterized by free-wall hypokinesis with apical sparing (McConnell sign), a finding possibly useful in differentiating pulmonary embolism from other causes of right ventricular dysfunction.32-38 Finally, echocardiography can be used to visualize massive pulmonary embolism directly in some patients.34 Therefore we believe that echocardiography is a practical and readily available diagnostic tool that should be considered in the evaluation of patients with suspected pulmonary embolism.
The typical echocardiographic findings include a normally contracting left ventricle, often with end-systolic obliteration of the LV cavity; a thin-walled, dilated, poorly contracting RV; right atrial enlargement; tricuspid insufficiency with a high-velocity regurgitant jet; increased estimated Pa pressures; leftward shift of the interventricular septum causing the typical “D” shape of the LV on the short-axis view (Fig. 38-4); paradoxical septal motion in systole; right Pa dilation; or loss of respirophasic variation in the inferior vena cava.39 RV infarction can usually be readily distinguished from acute pulmonary hypertension in that high Pa pressures are lacking. Right ventricular diastolic dimensions can be obtained by measuring right ventricular end-diastolic area in the long axis, from an apical four-chamber view, or by a transesophageal approach in the volume-repleted patient.40
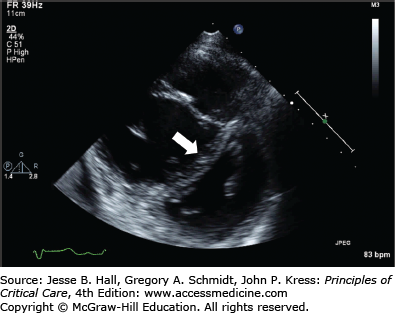
Enhanced echo techniques that are independent of geometrical assumptions have been developed to assess acute pathophysiological changes in RV function. Tricuspid annular plane systolic excursion (TAPSE),41 RV systolic and diastolic tissue Doppler imaging (TDI) velocities and Speckle tracking-derived strain42 TAPSE has been demonstrated to be a sensitive marker of acute RV dysfunction in 40 patients with acute PE.41
Newer algorithms for assessment of RV volumes and ejection fraction by real-time three-dimensional TTE are reported to be reasonably accurate. However in a meta-analysis of 23 studies that included 807 patients, 3D TTE underestimated RV volumes and EF when compared with the gold-standard measurements by cardiac MRI.43
Pulmonary Artery Catheterization: Pulmonary artery catheterization can estimate pulmonary arterial pressures more accurately than echocardiography. However, interpretation of mean pulmonary pressures and measurement of tricuspid regurgitation by thermodilution are confounded by technical limitations. RV failure is characterized by a reduced cardiac output (typically cardiac index <2.5 L/min/m2) and an elevation in right sided filling pressures (eg, right atrial pressure >8 mm Hg). A pulmonary artery catheter (PAC) with a fast-response thermistor has been advocated for accurate measurement of right ventricular end-diastolic volume (RVEDV) and hemodynamic parameters including RV ejection fraction by thermodilution in the presence of tricuspid regurgitation. However the fast-response thermistor PAC may systematically overestimate RVEDV in the presence of ischemia44 and has not been demonstrated to confer an improvement in survival.
Circulating Biomarkers: The utility of cardiac biomarkers for diagnosing acute RV injury in RHS has been demonstrated (mainly in acute PE) to accurately identify low-risk patients. BNP assay negative predictive values for in-hospital death range from 97% to 100%. RV systolic failure is an independent determinant of serum levels of brain natriuretic peptide (BNP) in patients with severe heart failure.35 However, the performance characteristics (positive predictive value and sensitivity) are inconsistent and preclude the use of either BNP or BNP levels across as range of cutoff values for routine diagnosis or prognosis in patients with moderate to high pretest probability.5
SPECIFIC RIGHT HEART SYNDROMES
Acute pulmonary hypertension is caused by an abrupt increase in pulmonary vascular resistance due to vascular obstruction or surgical resection. The prototype of acute pulmonary hypertension is acute pulmonary embolism (PE; see Chap. 39), but other forms of embolism (eg, air or fat), microvascular injury (eg, ARDS), drug effect, and inflammation can acutely raise pulmonary vascular resistance (see Table 38-3). In its most severe form, acute pulmonary hypertension associated with profound RV dysfunction is termed acute cor pulmonale.11,45,46 The echocardiographic diagnosis of acute cor pulmonale consists of the combination of RV dilation (reflecting RV diastolic overload) with paradoxical septal motion during systole (reflecting RV systolic overload).32
Right Ventricular Infarction: Right ventricular infarction is a well-recognized and fatal feature of inferior myocardial infarction.47,48 It is also seen in anterior infarcts. In most cases RV free wall infarction or ischemia is accompanied by varying degrees of septal and posteroinferior left ventricular injury, but relatively isolated RV injury is occasionally seen. RV myocardial injury and dysfunction representing noninfarcted hibernating myocardium may be able to sustain long periods of low coronary oxygen delivery and ultimately recover substantial contractile function.49
RV dilation accompanies significant myocardial injury. Concomitant LV infarction involving the interventricular septum may lead to further hemodynamic deterioration in patients with RV infarction because of the loss of LV septal contraction, which can assist RV ejection. Elevation of right atrial pressure on physical examination or direct measurement in a patient with an inferior myocardial infarction and clear lungs by exam and chest x-ray should lead to suspicion of RV infarction. When these features occur in a critically ill patient, the essential distinction is between RHS resulting from acute Pa hypertension and RHS resulting from RV infarction. Confirmatory evidence includes a right precordial electrocardiogram or echocardiographic evidence of RV injury (see Chap. 37). Proximal RCA occlusion commonly results in concomitant right atrial ischemia. This can precipitate significant rate and rhythm disturbances leading to further RH dysfunction and potentially AV dyssynchrony.49,50 Afferent vagal stimulation and baroreceptor stimulation in ischemic myocardial tissue lead to enhanced parasympathetic tone and the cardioinhibitory Bezolde-Jarisch reflex.51 Reperfusion therapies can paradoxically exacerbate this response resulting in severe but transient bradycardic hypotension.
Meta-analysis of 22 studies involving 7136 patients with AMI identified 27.5% with RV myocardial involvement. The pooled relative risk mortality increase for RVI patients was 2.59 (95% CI, 2.02–3.31; Z = 7.57; p <.00001) compared with non-RV MI.52 The mortality risk was consistent across studies where RVI was diagnosed by ECG alone or supplemented by echocardiographic confirmation. RVI was significantly more frequently complicated cardiogenic shock, ventricular arrhythmias, and mechanical complications including papillary muscle rupture.
The focus of management in RV infarction is on maintenance of optimal RV preloading to avoid worsened RV distension, preservation of RV synchrony, reduction in RV afterload (particularly when LV dysfunction is present), and inotropic and mechanical support of the RV.53
Early reperfusion with fibrinolytics or direct coronary intervention has been advocated based on clear evidence of early reperfusion,54 functional RV performance improvement and improved hospital mortality. However, even delayed efforts at reperfusion with fibrinolytic or directed PCI are indicated for most patients. Echocardiography can be highly useful in confirming RV infarction and in determining the response to therapeutic interventions.
Acute Lung Injury/Acute Respiratory Distress Syndrome: As many as one quarter of patients with ARDS develop significant pulmonary hypertension and RV dysfunction45,46 although the frequency of this complication in an era when Pa catheters are infrequently inserted is likely to be clinically underappreciated. Contributors to Pa hypertension in ARDS include hypoxic pulmonary vasoconstriction, mediator release, high alveolar pressure during mechanical ventilation, and microthrombi in situ.
In retrospective analysis of 145 patients from the French Pa Catheter study, Osman and coworkers identified right ventricular failure in 9.6% of patients defined as a mean PAP >25 mm Hg, CVP > PAOP and stroke volume index <30 mL/m2, although there was no independent association with 90-day survival in that study. Right ventricular dysfunction characterized by PRA > Ppao has been established to be an early independent predictor of mortality in ARDS (OR 5.1 95% CI 1.5-17.1; P = 0.009).55 The large prospective European ARDS Collaborative Study evaluated pulmonary hemodynamic variables in 424 of 586 ARDS patients.56 In most patients, mean Pa pressure was modestly elevated on admission (26.1 ± 8.5 mm Hg) and was persistently elevated at 48 hours in nonsurvivors compared with survivors (28.4 ± 8.5 mm Hg vs 24.1 ± 6.7 mm Hg). The ratio of RV to LV stroke work was also significantly elevated in all patients, and along with the ratio of partial oxygen pressure to the fraction of inspired oxygen (OR 0.96-0.98), was identified as an independent predictor of survival (OR 20-85; p = 0.0001). In a retrospective analysis of the ARDS Network, Fluid and Catheter Treatment Trial dataset, Bull, et al identified a high frequency of pulmonary vascular dysfunction and a strong association between an increased transpulmonary gradient at enrollment (TPG; Pa mean pressure—Pa occlusion pressure ≥12 mm Hg) with increased 60-day mortality rate (30% vs 19%; P = 0.02). Notably if the TPG failed to normalize by day 5, this too was associated with increased likelihood of death (36% vs 19%; P = 0.01).
Shunting through a patent foramen ovale as a consequence acute pulmonary hypertension with RV dysfunction in ARDS is common and is associated with prolonged ventilator dependence. 39 out of 116 sequential ARDS patients in one French prospective cohort had PFO-associated shunt demonstrated by bubble-contrast TEE.46
These findings would suggest an aggressive approach to lowering RV afterload in patients with ARDS by reducing alveolar pressures and administering inhaled nitric oxide or prostacyclin. However, despite reproducible reductions in Pa pressure and improvements in oxygenation indices, randomized controlled studies using this approach have repeatedly failed to demonstrate a survival benefit, as discussed below.
Mechanical ventilation in the prone position (PPV; reviewed in Chap. 52) has consistently been demonstrated to improve gas exchange in adults with ARDS.57 However, whether PPV improves survival in unselected ARDS patients is unresolved. PPV has been demonstrated to exert it’s salutary effects by reducing antero-posterior pleural pressure gradients, reducing the pro-atelectatic pressure effects of the heart and ventral trans-diaphragmatic pressures and reducing heterogeneity of regional lung V/Q relationships.58 As a consequence of these mechanisms, PPV has also been demonstrated to unload the RV in patients with ARDS and acute cor pulmonale.59 In 21 patients with severe ARDS and acute pulmonary hypertension with RV dysfunction, 18 hours of PPV significantly decreased end-inspiratory plateau pressure and PaCO2 despite unchanged tidal volumes. Additionally PPV was associated with a significant improvement in RV function compared with SPV as assessed by right ventricular enlargement and septal dyskinesia at transesophageal echocardiography.
Sepsis: Sepsis itself is probably capable of causing pulmonary hypertension, even in the absence of acute lung injury, based on animal models60 and limited human studies.61,62 Although common in patients with severe sepsis, it is our experience that acute pulmonary hypertension is only of clinical importance when ARDS (or another clear precipitant) is present. Sepsis-associated proinflammatory cytokines particularly tumor necrosis factor-α have been demonstrated to induce caspase-mediated apoptosis and myocyte dysfunction63 as well as having negative inotropic effects on the ventricular myocardium. It seems likely that the systemic hypotension of septic shock also renders the RV more vulnerable to ischemic systolic dysfunction when combined with modest increases in afterload.64 It has been argued that this right ventricular perfusion gradient accounts for the differentially impaired perfusion and contractility of the RV compared with the LV in sepsis.62
A notable insight into the complex role of endogenous nitric oxide in regulating pulmonary vascular tone in septic shock patients was derived from a randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88.65 Patients who were randomized to the treatment arm had a 10% absolute higher mortality rate at 28 days than patients in the placebo arm. 546C88-treated patients had a greater incidence of pulmonary hypertension, with an initial increase in the pulmonary vascular resistance and a sustained reduction in the pulmonary venous admixture, possibly through augmented hypoxic pulmonary vasoconstriction. Three patients in the treatment arm developed right heart failure. It has been suggested that sepsis-associated NO production may have a partially protective effect on the pulmonary vasculature by optimizing pulmonary ventilation-perfusion relationships.
Acute Sickle Chest Syndrome: Acute pulmonary hypertension complicates vaso-occlusive sickle cell crises66 and can present as the acute sickle chest syndrome (ACS). ACS results from pulmonary microvascular in situ thrombosis, pulmonary fat embolism from infarcted long bone marrow, and hypoxic vasoconstriction. Recurrent episodes result in secondary chronic pulmonary hypertension and cor pulmonale. Amongst 70 patients experiencing 84 ACS crises, acutely elevated pulmonary arterial pressures (estimated by echo tricuspid regurgitant jet velocity, TRV) was present in 60% (37% of patients had a TRV >3.0 m/s) and was associated with RV dilation in most patients and RV systolic failure (cor pulmonale) in 13%.67 Acute Pa hypertension generally resolved after ACS resolution (PASP decreased from 51 [47-67] mm Hg during severe ACS to 25 [35-40] mm Hg [P < 0.01]). However, severe pulmonary hypertension (TRV >3.0 m/s) during ACS was associated with a dramatic reduction in long-term survival; only 50% of those patients survived to 36 months versus 100% of patients with TRV <3.0 m/s; P = 0.007.
Related posts:






Full access? Get Clinical Tree


