79 Ventricular Arrhythmias
 Normal Electrophysiology
Normal Electrophysiology
Anatomic Synopsis
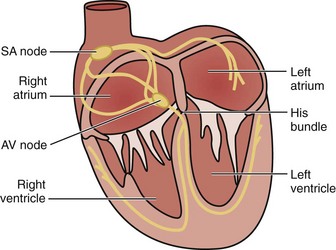
The cardiac electrical impulse originates in the sinoatrial (SA) node, located high on the right atrium near its junction with the superior vena cava (Figure 79-1). The impulse then propagates through muscle fibers and specialized internodal pathways (composed of Purkinje-type fibers) to converge on the AV node, located in the interatrial septum near the tricuspid valve and the opening of the coronary sinus. From the AV node, the impulse travels through the bundle of His, its left and right branches, and the Purkinje system to simultaneously activate the right and left ventricles. A ring of fibrous tissue interposed between the atria and the ventricles prevents spread of the electrical impulse through the muscle fibers, enabling the AV node to function as a relay and filter structure limiting the number of impulses that can be transmitted to the ventricles and thus maintaining the ventricular rate within a range that is physiologically permissible for stroke volume generation. Thus, the AV node prevents a 1 : 1 conduction under conditions of very rapid atrial activation such as atrial flutter (rate ≈ 300 s−1) and atrial fibrillation (rate ≈ 350 s−1 to 600 s−1), typically preventing increases in ventricular rate above 150 s−1 or 180 s−1 in instances of atrial flutter or atrial fibrillation, respectively.
Action Potential and Pacemaker Activity
Action potential results from rapid depolarization and repolarization of polarized cells driven by a coordinated sequence of opening and closing of channels that regulate ion currents across the cell membrane. The main ion currents are carried by channels that regulate influx of sodium ions (Na+) and calcium ions (Ca2+) (inward currents) and by efflux of potassium ions (K+) (outward currents).1–3
The action potential is essential to initiate and propagate the electrical impulse throughout the conduction system and ultimately reach cardiomyocytes where the action potential signals activating of contractile activity and therefore pump function through Ca2+-induced Ca2+-release from the sarcoplasmic reticulum. Accordingly, the electrical impulse typically precedes mechanical activity. However, cardiomyocytes can also react to mechanical forces through stretch-activated ion channels and other related mechanisms including mechanical modulation of Ca2+ handling and interaction with other mechanosensitive cells in the heart.4,5 This mechanism is in part responsible for commotion cordis,6,7 precordial thump,8 and fist pacing.9
The functional characteristics of the action potential differ contingent on the cell type. Cells from the Purkinje system and from atrial and ventricular muscle are primarily responsible for propagation of the action potential. These cells have a stable resting potential at approximately −90 mV (inside negative), which is largely the result of a K+ current known as the inward rectifier (IK1). IK1 “anchors” the membrane potential to a voltage close to the equilibrium potential of K+;2 it is turned off during depolarization (inward rectification). Initiation of an action potential requires depolarization of the membrane potential between −70 and −80 mV. This voltage is the threshold at which fast voltage-gated Na+ channels are activated, prompting Na+ influx driving an inward current (INa).3 Depolarization to the threshold potential typically occurs upon arrival of an action potential. The INa drives the membrane potential toward the equilibrium potential of Na+, causing further depolarization and reversal of the membrane potential to approximately +20 mV (overshoot). This phase is known as phase 0 of the action potential and ushers in a 4-phase repolarization (Figure 79-2). Phase 1 is the initial early repolarization (action potential notch) and results from rapid inactivation of Na+ channels (inner gate) and the opening of K+ channels carrying a rapidly activating and rapidly inactivating “transient” outward current (ITo). Based on recovery time, two distinct subpopulations of channels can be recognized carrying a rapidly “fast” recovering current (ITof) and a slowly recovering current (ITos). The difference in these currents rely on distinct pore-forming α-subunits with Kv4.2/Kv4.3 (KCND2/KCND3) genes encoding ITof and Kv1.4 encoding ITos.10 It appears that ITof is the predominant contributor to ITo in ventricular myocardium.11 Because the K+ channels carrying ITo are expressed in the subepicardial and mid-myocardial regions but not in the subendocardial region, they contribute to the inhomogeneity of repolarization.12
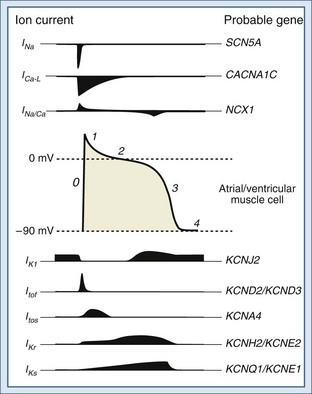
Phase 2 is the plateau phase of the action potential and results mainly from a Ca2+ current carried by the slow and prolonged opening of L-type voltage-gated Ca2+ channels (ICa-L).13,14 Opening of these channels begins during phase 0 at a membrane potential of −30 to −40 mV. These channels are inactivated in response to increases in cytosolic Ca2+ and are strongly regulated by neurotransmitters. Phase 3 corresponds to late repolarization and follows the closing of Ca2+ channels and opening of K+ channels with slow activation kinetics carrying currents known as delayed rectifiers (IK). The IK are the main repolarizing currents and have two components carried by distinct gene products: a rapid component (IKr) and a slow component (IKs).15,16 Both are implicated in the heritable forms of long QT syndrome (see later discussion).17 In addition, opening of IK1 (main contributor to the resting membrane potential as described earlier) contributes to repolarization. Phase 4 represents return to the resting membrane potential and the interval during which ionic balance is restituted, largely through the action of the Na+/K+ pump.
Cells of the SA and AV nodes lack voltage-gated Na+ channels, and phase 0 is carried by ICa-L.18 Because of their slower opening kinetics (relative to Na+ channels), they give rise to a slanted phase 0 and in part determine the lower conduction velocity of the SA and AV nodes (≈50 cm·s−1) compared with the His-Purkinje system (≈400 cm·s−1) and muscle cells (≈100 cm·s−1). SA and AV node cells also have pacemaker activity and slowly depolarize during phase 4 to a threshold potential of approximately −40 mV. The slow depolarization is called pre-potential or pacemaker potential and involves a background Na+ current (INa-B), a decay of K+ currents, and the opening of T-type voltage-gated Ca2+ channels (ICa-T) at a potential between the thresholds for INa and ICa-L, unleashing phase 0. Cells of the His-Purkinje system have latent pre-potential activity and can become active when SA or AV node activity is depressed or their impulse is blocked. Atrial and ventricular muscle cells exhibit pre-potential activity only under abnormal circumstances (see later discussion).
The preceding description is succinct and oversimplified. Various other ion channels, antiporters, pumps, and receptors play important roles in specific physiologic states and disease processes. For example, there is a nonselective cationic channel that is gated at resting potential by intracellular Ca2+ and produces an inward Na+ current (INS).19 This current may contribute to delayed afterdepolarizations following Ca2+ release by the sarcoplasmic reticulum. Ik(atp) is a K+ current carried through metabolically regulated channels that are inhibited by adenosine triphosphate (ATP) and opened under conditions of ischemia and hypoxia. Ik(atp) is the main contributor to the shortening of the action potential duration and the characteristic ST-segment elevation observed in the surface electrocardiogram during myocardial ischemia.20,21
The sarcolemmal Na+/Ca2+ exchanger is another important modulator of the action potential. Because it exchanges one Ca2+ for three Na+, it is electrogenic and generates a current (INa/Ca) whose direction is determined by the Na+ and Ca2+ gradients and the membrane potential.21,22 In settings in which there is cytosolic Ca2+ overload (e.g., ischemia and reperfusion, digitalis toxicity), Ca2+ may trigger Ca2+ release from the sarcoplasmic reticulum during phase 4, which in turn prompts reverse-mode operation of the Na+/Ca2+ exchanger, causing an inwardly directed INa/Ca (Na+ influx). This current contributes to the generation of delayed afterdepolarizations and triggered arrhythmias (see later discussion).
Adrenergic receptors also play important roles in modulating the action potential by modifying channel activity.23–25 For example, stimulation of β-adrenergic receptors increases the activity of ICa-L, leading to increased Ca2+ influx, signaling increased contractile activity. β-Adrenergic receptor stimulation can also activate K+ channels, shortening repolarization and the duration of the action potential.26 α1-Adrenergic receptors exert actions via G-protein on the Na+/K+ pump, K+ channels, and phospholipase C and can alter impulse initiation and repolarization. α1-Adrenergic stimulation has been linked to triggered rhythms via early and delayed afterdepolarizations and the development of abnormal automatic rhythms in the setting of ischemia and reperfusion.27,28
 Mechanisms of Ventricular Tachyarrhythmias
Mechanisms of Ventricular Tachyarrhythmias
Abnormalities in Impulse Generation
Abnormalities in impulse generation are generally the result of automaticity or triggered activity.
Automaticity
Abnormal automaticity refers to the generation of impulses in cells that normally do not exhibit pacemaker potential. This phenomenon can occur in cells that are partially depolarized as a result of a pathologic process (e.g., ischemia). Under these conditions, the reduction in the resting membrane potential (less negative; to −70 or even −50 mV) shifts the balance during phase 4 toward depolarizing currents.29 Through this mechanism, automaticity can developed in atrial and ventricular muscle cells and in specialized tissues other than the SA in which the firing conditions can be altered. Examples of abnormal automaticity include accelerated idioventricular rhythms and some VTs that develop 24 to 72 hours after an acute myocardial infarction.30,31
Triggered Activity
Triggered activity refers to arrhythmias that arise from afterdepolarizations. Afterdepolarizations are alterations in membrane potential that occur during the repolarization phase without intervening external triggers or cell-to-cell interactions.32 Afterdepolarizations can develop in different phases of the action potential. Those that develop during phase 2, phase 3, or early phase 4 and are called early afterdepolarizations and are characterized by transient retardations in repolarization with or without upturn of the membrane potential (Figure 79-3). An upturn of sufficient magnitude may trigger an “extra” action potential before the cycle is over. Early afterdepolarizations are typically associated with conditions that prolong the action potential duration, such as decreased inactivation of fast INa (e.g., long QT3 syndrome) or decreased outward K+ currents (e.g., IKs in long QT1 and IKr in long QT2 syndromes) prolonging Ca2+ entry through ICa-L.33 The development of early afterdepolarizations in this setting is thought to trigger torsades de pointes. Early afterdepolarizations are also associated with increased sympathetic tone, use of catecholamines, hypoxia, acidosis, and bradycardia.
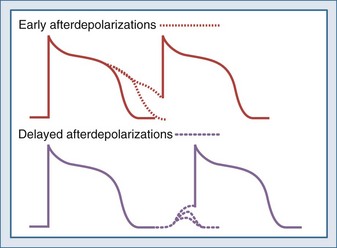
Afterdepolarizations that occur in late phase 4 are called delayed afterdepolarizations and are characterized by low-amplitude depolarizations that may reach threshold and trigger an action potential (see Figure 79-3). The main underlying abnormality in delayed afterdepolarizations is intracellular Ca2+ overload, promoting Ca2+ release from the sarcoplasmic reticulum33 and depolarizing currents (i.e., inward INa/Ca currents). Delayed afterdepolarizations are classically associated with digitalis toxicity; however, many other conditions favoring cytosolic Ca2+ overload can also produce delayed afterdepolarizations such as myocardial stretch, hypertrophy, catecholamines, ischemia, and reperfusion. In the setting of heart failure, increased expression of Na+-Ca2+ exchanger along with abnormalities in the ryanodine receptor has been shown to predispose to delayed afterdepolarizations.
Abnormalities in Impulse Conduction (Reentry)
Abnormalities in impulse conduction account for the vast majority of sustained ventricular tachyarrhythmias consequent to a phenomenon known as reentry. Reentry occurs when a normally propagating impulse reenters a region of previously excited tissue after its refractory period is over and excites it again. Reentry can continue to repeat, originating a tachyarrhythmia. Several forms of reentry have been described, including circus movement, phase 2, and reflection.34
Circus Movement
The ring model is the simplest of all35 and can be used to illustrate the basic mechanism of reentry (Figure 79-4). The ring model requires two anatomically contiguous paths in specialized tissue or in muscle fibers separated by a central area of unexcitable tissue. One of these paths (b in Figure 79-4) must exhibit a zone of unidirectional block allowing the impulse to propagate in only one direction. The alternative path (a in Figure 79-4) allows the impulse to circumvent the unidirectional block. Conduction through this alternative path should be slow or refractoriness proximal to the path should be brief to allow recovery of excitability. Once the impulse reaches the distal end of the alternative path, it propagates in a retrograde manner through the path of unidirectional block to reenter the proximal end of the alternative path. For the cycle to repeat (and establish a reentry circuit causing tachyarrhythmia), the wavelength of the circling impulse must be shorter than or at least equal to the length of the reentry circuit (or path length), thus enabling the leading edge of the circling impulse to find the tissue in an excitable state. The wavelength of the circling impulse is defined as the product of conduction velocity and duration of the refractory period.
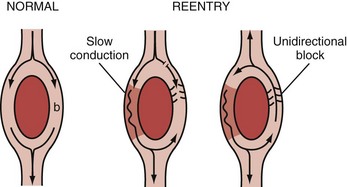
The leading circle model is similar to the ring model but does not require anatomic obstacles and can develop in structurally uniform myocardium by a properly timed premature impulse.36,37 The figure-of-eight model was first described in experimental myocardial infarction. It refers to two reentry circuits moving alongside a functional conduction block (ischemia or infarct) in opposite directions, forming a pretzel-like configuration.38 The spiral wave model is considered a more complex version of the leading circle model. It involves a core and filaments and is usually described as reentry in two dimensions.39,40 The spiral wave model has been used to explain the electrocardiographic patterns associated with monomorphic and polymorphic VTs and also VF. In monomorphic VTs, the spiral wave is thought to be anchored and unable to drift within the myocardium, whereas in polymorphic VTs, such as torsades de pointes, the spiral is thought to drift. In the case of VF, the spiral wave is believed to break up into multiple rotating spiral waves that are continuously extinguishing and recreating. However, some authors have proposed a single rapidly shifting spiral, and others have postulated a stationary rotor whose frequency of excitation is exceedingly high, resulting in multiple areas of intermittent block.41
Phase 2
Phase 2 reentry refers to the generation of local reexcitation as a result of increased heterogeneity of repolarization. This phenomenon occurs when repolarization is markedly shortened in certain regions of the myocardium—essentially obliterating phase 2 of the action potential (plateau phase)—but is maintained in others. This creates conditions conducive to local reexcitation, which may precipitate ventricular tachyarrhythmias during myocardial ischemia.42 During ischemia, action potentials of normal duration alternate with ones of shorter duration, yielding beat-to-beat alternans (temporal dispersion) and site-to-site alternans (spatial dispersion) and promoting regions with conduction block and regions with injury current, leading to reentry and ventricular tachyarrhythmias. The degree of spatial and temporal dispersion progresses along with the duration of ischemia, suggesting that this mechanism may be an important trigger of VT and VF during acute myocardial ischemia.43,44 In the surface ECG, dispersion of the action potential duration manifests as T-wave alternans, which is a predictor of VF.45
Reflection
Reflection refers to a back-and-forth propagation of the impulse over the same functionally unexcitable tissue, with recurrent activation of the proximal region as a result of electrotonic currents.46,47 The area of unexcitable tissue could result from ischemia and lead to extrasystolic activity. Reflection differs from classic reentry in that the impulse travels along the same pathway in both directions.
 Conditions Predisposing to Ventricular Arrhythmias
Conditions Predisposing to Ventricular Arrhythmias
Channelopathies
The term “channelopathies” has been coined to identify a group of diseases characterized by abnormalities in the proteins that form ion channels.48,49 These abnormalities distort the normal action potential, primarily accentuating the inherent instability of repolarization and increasing the risk of polymorphic VT of the torsades de pointes type. Channelopathies may be hereditary or acquired.
Hereditary Channelopathies
Long QT Syndrome
The vast majority of hereditary channelopathies result from mutations in genes that encode for Na+ and K+ channels, with the most representative being the long QT syndrome.50–52 The long QT syndrome was first described in 1957 by Jervell and Lange-Nielsen in a group of patients with long QT intervals, episodes of torsades de pointes, and deafness.53 This syndrome is transmitted by autosomal recessive inheritance and is known as the Jervell and Lange-Nielsen syndrome. In 1963 and 1964, Romano and colleagues54 and Ward55 independently reported patients with an almost identical disorder but without deafness. This syndrome is transmitted by autosomal dominant inheritance and is known as the Romano-Ward syndrome.
It is now recognized that long QT syndrome results from mutations in at least 12 genes, leading to distinct types designated long QT1 through long QT12 (Table 79-1). Long QT1 is the principal genetic type responsible for both Jervell and Lange-Nielsen and Romano-Ward syndromes and accounts for nearly 50% of all genotyped families. Long QT2 accounts for nearly 40% and long QT3 for about 5%. The remaining types are much less frequent.56
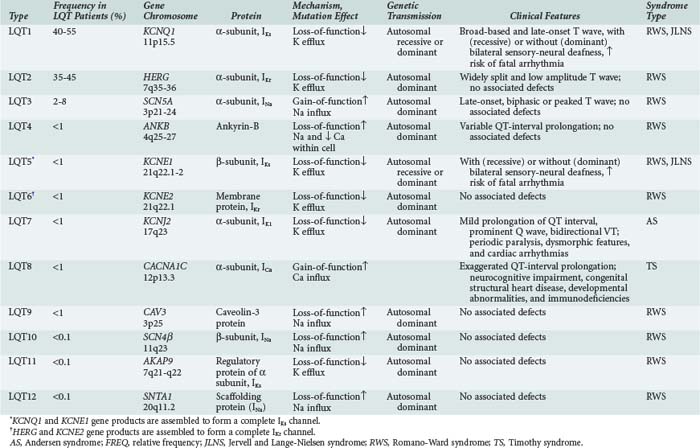
Long QT3 stems from a mutation in SCN5A, the gene that encodes the α-subunit of the fast cardiac Na+ channel. SCN5A mutation causes a gain of function leading to incomplete channel inactivation and persistence of INa during the plateau phase of the action potential.57,58
Long QT4 has been linked to a loss-of-function mutation in the ANKB gene.59 This gene encodes ankyrin-B, which is a member of a family of versatile membrane adapters. Ankyrin-B, among other functions, coordinates the opening and closing of calcium, potassium, sodium, and chloride channels. The failure to properly coordinate the opening and closing of ion channels leading to long QT syndrome and arrhythmias illustrates a novel mechanism of arrhythmias.
In long QT9, QT10, and QT12, the mutations affect the genes CAV3, SCN4β, and SNTA1, leading to a gain of function in INa, resulting in increased Na+ influx within the cardiac cell.60,61
Accordingly, the common mechanistic thread among long QT syndromes is perturbation of the balance between INa and IK during the plateau phase of the action potential, yielding prolongation of repolarization, a reduced rate of ICa-L inactivation, late Ca2+ influx, and early afterdepolarizations predisposing to torsades de pointes.62
The diagnosis is suspected in young individuals who present with syncope or episodes of sudden death typically during exercise, emotional distress, or exposure to factors that cause prolongation of the QT interval. A family history of unexplained syncope or sudden cardiac death, especially in young kindred, should raise suspicion. Sudden cardiac death occurs in approximately 4% of affected individuals. The diagnosis should be suspected when the corrected QT interval (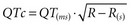 ) exceeds 470 milliseconds in males (normal <422 milliseconds) and 480 milliseconds in females (normal <432 milliseconds) in the absence of other conditions that may lengthen the QT interval. In addition, there may be sinus bradycardia with sinus pauses in about one-third of individuals (especially in long QT3), QT dispersion, and various T-wave abnormalities (e.g., notched, bifid, biphasic). Factors predisposing to sudden cardiac death include recurrent syncope, survival from cardiac arrest, congenital deafness, female sex, relative bradycardia, corrected QT interval greater than 600 milliseconds, and kinship with a symptomatic patient.50 Genetic testing for identifying the various long QT subtypes is becoming readily available, with its impact on risk stratification and guidance for placement of an implantable cardioverter-defibrillator (ICD) to be determined by future research.56,60
) exceeds 470 milliseconds in males (normal <422 milliseconds) and 480 milliseconds in females (normal <432 milliseconds) in the absence of other conditions that may lengthen the QT interval. In addition, there may be sinus bradycardia with sinus pauses in about one-third of individuals (especially in long QT3), QT dispersion, and various T-wave abnormalities (e.g., notched, bifid, biphasic). Factors predisposing to sudden cardiac death include recurrent syncope, survival from cardiac arrest, congenital deafness, female sex, relative bradycardia, corrected QT interval greater than 600 milliseconds, and kinship with a symptomatic patient.50 Genetic testing for identifying the various long QT subtypes is becoming readily available, with its impact on risk stratification and guidance for placement of an implantable cardioverter-defibrillator (ICD) to be determined by future research.56,60
In addition to long QT syndromes, recent population studies63 have shown that even milder prolongation of the QTc in adults (>450 milliseconds in men and > 470 milliseconds in women) increases the risk of sudden cardiac death by threefold after adjustment for age, gender, body mass index, hypertension, cholesterol/high-density lipoprotein ratio, diabetes mellitus, myocardial infarction, heart failure, and heart rate. In the same population, QTc prolongation was strongly associated with variant rs10494366 T>G and rs10918594 C>G of the nitric oxide synthase 1 adaptor protein (NOS1AP) gene.64 More recent studies show that use of the calcium channel blocker, verapamil, in patients with these variants prompts a greater QTc prolongation than in patients with the wild genotype.65
Short QT Syndrome
Short QT is a more recently described syndrome66 characterized by tall and peaked T waves with QT interval ≤ 300 milliseconds, insensitive to changes in heart rate, and a structurally normal heart. Individuals are at risk of developing VF and also atrial fibrillation and may complain of palpitations and episodes of syncope. They may also have family members with a similar history or a history of unexplained or sudden death at a young age.
Brugada Syndrome
Another important hereditary channelopathy is Brugada syndrome, described in 1992 by the Brugada brothers,67–70 who noticed an association between sudden cardiac death and ST-segment elevation in V1 to V3, with a pattern resembling right bundle branch block in individuals with structurally normal hearts.
Brugada syndrome results in part from mutations in the SCN5A gene (encoding for the INa α-subunit). In contrast to long QT3—which also affects the SCN5A gene—the mutations in Brugada syndrome lead to a loss of function resulting in accelerated inactivation of INa during phase 1, leaving the repolarizing ITo current unopposed and consequently prompting rapid repolarization and shortening of the action potential duration (refer to Figure 79-2 for the INa and ITo contributions to the action potential). Because ITo is expressed predominantly in the epicardium, the normally depolarized endocardium can re-excite the prematurely repolarized epicardium, leading to phase 2 reentry and generation of a phase 2 reentrant premature beat that could capture a vulnerable window and precipitate VT (which is typically polymorphic) and/or VF.
Brugada syndrome exhibits predominantly an autosomal dominant pattern of inheritance, with an average worldwide prevalence of 5 : 10,000. More than 100 mutations in seven genes have been identified. Loss-of-function mutations in the SCN5A gene cause approximately 20% of cases. A few mutations have been described in the GPD1L gene, which encodes the glycerol-3-phosphate dehydrogenase-1 like protein; the CACNA1C gene, which encodes the α-subunit of the Ca(v)1.2 ion channel conducting the inward current (ICa-L); the CACNB2 gene, which encodes the β2-subunit of the Ca(v)1.2 ion channel; the SCN1B and SCN3B genes, which in the heart encode the β-subunits of the Na(v)1.5 sodium ion channel; and the KCNE3 gene, which encodes the ancillary inhibitory β-subunit of several potassium channels including the Kv4.3 ion channel conducting the repolarizing potassium current ITos.71
The ST-segment elevation can adopt various shapes that have been related to the severity of the INa/ITo imbalance, including—in order of increasing severity—saddleback, coved, and triangular shapes.62 These changes are dynamic and can change in the same affected individual as shown in Figure 79-5.
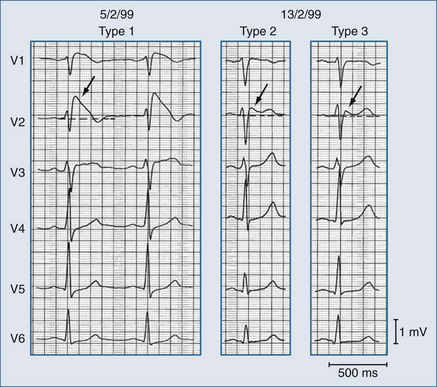
Brugada syndrome is the major but not the only cause of sudden unexpected death syndrome (SUDS)72,73 and is the most common cause of sudden death in young men without known underlying cardiac disease in Thailand and Laos.74 However, Brugada syndrome exhibits variable expressivity, reduced penetrance, and “mixed phenotypes” where families may contain members with Brugada syndrome as well as members with short QT syndrome, long QT syndrome, atrial fibrillation, disease of the conduction system, and even structural heart disease.71
Patient with Brugada syndrome may have concealed or intermittent forms that can be unmasked (or precipitated) by febrile states, vagotonic agents, α-adrenergic agonists, β-adrenergic blockers, tricyclic or tetracyclic antidepressants, a combination of glucose and insulin and hypokalemia, and alcohol and cocaine toxicity.75
A useful clinical test to identify concealed Brugada syndrome is the administration of class IC antiarrhythmic drugs: Na+ channel blockers such as ajmaline (1 mg/kg intravenous (IV) in 5 minutes), flecainide (2 mg/kg IV in 10 minutes), or procainamide (10 mg/kg IV in 10 minutes). The test should be performed in an environment with capability for immediate defibrillation, because there is a 0.5% risk of precipitating VF. Of the drugs listed above, ajmaline is the preferred drug because of its very short half-life.76 The test is considered positive if an additional 1-mm ST-segment elevation (measured 0.08 seconds after the J point) occurs in leads V1, V2, and V3. The test is highly specific and should be considered in patients who present with history of syncope of unknown origin or with idiopathic VF.
Patients with the Brugada syndrome must be treated with an ICD, because antiarrhythmic agents have not been found in general to be effective.69 However, recent studies have shown that quinidine and hydroquinidine can prevent spontaneous ECG changes and reduce the risk of VT and VF, presumably through inhibition of ITo.77,78 Although such a pharmacologic approach is currently regarded as an adjunct to ICD placement to reduce the number of shocks, it could be considered in high-risk patients when ICD placement is not possible.



Full access? Get Clinical Tree


