
FIGURE 97.1 Mid esophageal aortic valve short axis (A) and long axis (B). TEE views showing calcified aortic valve leaflets with valve stenosis.
Pathophysiology
In chronic AR, the LV dilates and hypertrophies due to volume overload. This keeps wall stress in check and maintains normal forward stroke volume. As the disease progresses and the compensatory limit is reached, the wall stress begins to rise and systolic function deteriorates, with decreasing forward stroke volume as well as increasing LV end-diastolic volume (LVEDV) and LVEDP, resulting in heart failure. During the relatively asymptomatic phase, the patient develops symptoms at a rate of 3.7% per year. The presence of symptoms, systolic dysfunction, and an increase in end-systolic dimensions indicate severe and decompensated AR.
| TABLE 97.2 Echocardiographic Assessment of Aortic Stenosis (AS) | |||
 | |||

FIGURE 97.2 Spectral Doppler through the aortic valve showing aortic valve gradients and calculated aortic valve area.
In acute AR, the LV is subject to a sudden increase in volume, with no opportunity for a compensatory increase in compliance and eccentric hypertrophy to occur. Therefore, AR of a lesser severity may markedly increase end-diastolic pressures, causing acute pulmonary venous congestion and pulmonary edema. Symptoms become more severe as the regurgitant orifice size increases, and with bradycardia as the duration of diastole increases.
Diagnosis
An ECG is performed to rule out ischemic heart disease in situations of acute AR. The chest radiograph may reveal cardiomegaly in chronic AR or pulmonary congestion with a normal-sized heart in acute AR. Echocardiography is essential to determine the mechanism and severity of AR, and identifies perivalvular pathology (e.g., an abscess) or associated aortic pathology such as acute aortic dissection. In the presence of normal leaflets, the aortic valve can be preserved surgically (AV repair). Diseased valve with vegetations, calcifications, and restrictions usually end up in AVR. Other techniques such as computerized tomographic (CT) scanning, magnetic resonance imaging (MRI), and aortography have been used. Each modality has its own advantages and disadvantages (5). TEE is clearly superior to MRI and CT scanning to characterize the valve pathology in cases of acute dissection, and obviates the need for aortography (Fig. 97.3). TEE is also readily available, can be done at the bedside and will avoid transport of hemodynamically unstable acute AR patients to radiology suite. TEE can also identify the dissection flaps causing coronary artery occlusion. Analysis of wall motion abnormalities may also help in ruling out coronary artery involvement in dissections. Coronary angiography may be necessary to rule out ischemic heart disease before surgery, in patients with chronic AR.

FIGURE 97.3 A: TEE images showing type A aortic dissection in the ascending aorta. B: Dissection flap in the aortic arch, with arrows showing the intimal flap. C: Type B aortic dissection in the descending aorta.
Treatment
While medical therapy may allow patients with mild acute AR to reach a chronic compensated state, patients with acute AR are generally ill enough to require ICU admission and/or emergency AVR. Patients with ascending aortic dissections and acute-onset AR will require immediate surgical management. In the absence of aortic dissection, the medical management of acute AR aims at optimizing CO and systemic perfusion, reduce pulmonary venous congestion, and treat any associated disorder. Invasive monitoring is initiated and intravascular volume is optimized. Moderate tachycardia is beneficial in maintaining CO, and decreases the regurgitant fraction by decreasing the duration of diastole. β-Blockers should be administered with careful hemodynamic monitoring because they depress cardiac function, increase the duration of diastole, and can precipitate circulatory failure. Hypertension and increased afterload are to be avoided; afterload reduction is indicated with arterial vasodilators such as nitroprusside. Inotropic therapy is advised only in patients with depressed systolic function. An IABP is absolutely contraindicated in patients with AR, as it will increase the regurgitant fraction.
In patients with chronic AR who present with an acute decompensation, a search should be made for the precipitating cause, with particular attention to possible infectious endocarditis. Most patients stabilize with medical therapy, but early elective surgery should be considered, as the outlook for medically treated symptomatic patients is poor. Decompensated patients who do not improve with aggressive medical therapy should undergo emergency valve replacement. Mortality with medical therapy alone in this group approaches 100%, while many moribund patients will survive with surgery.
In contrast to other causes of AR, inotropic therapy is avoided in patients with aortic dissection, as it occurs as a result of long-standing, poorly controlled hypertension or trauma. Left ventricular contractility is preserved and inotropes are not indicated. Moreover, β-adrenergic blockade is often necessary to reduce the velocity of LV ejection and aortic wall stress, therefore preventing extension of the aortic dissection or aortic rupture.
Mitral Stenosis
Mitral stenosis (MS) is mostly related to rheumatic heart disease. Degenerative calcific stenosis, congenital stenosis, and connective tissue disorders such as systemic lupus erythematosus and Lutembacher syndrome (atrial septal defect with MS) are other causes for MS. Atrial myxomas and left atrial ball-valve thrombus can present with intermittent obstruction to mitral inflow and mimic MS.
Pathophysiology
In rheumatic MS, inflammation of the connective tissue leads to leaflet thickening and calcification, and commissural and chordal fusion. Ultimately, many patients are left with a funnel-shaped mitral apparatus. As the condition progresses, left atrial pressure and the transmitral gradient increase, thus maintaining flow. However, the subsequent increase in pulmonary venous pressure leads to hydrostatic pulmonary edema. Pulmonary vasoconstriction increases pulmonary arterial pressure and right ventricular afterload. Over the course of rheumatic heart disease, persistently elevated pulmonary arterial pressure leads to intimal hyperplasia and medial hypertrophy. These structural changes are permanent and result in fixed pulmonary hypertension. Right ventricular dilatation and failure result in tricuspid insufficiency and systemic venous congestion, respectively. The impact of MS on CO is initially determined by the limitation to flow across the narrowed mitral orifice. However, as pulmonary hypertension progresses, right ventricular failure may become severe enough to limit CO. Similarly, progression of rheumatic disease with its associated inflammation, fibrosis, and calcium deposition may impair left ventricular function. Although this effect is not a major determinant of CO in patients with MS, it may become important to consider in some patients after surgical repair. In such situations, the increase in transmitral flow may expose the LV to a sudden increase in preload, causing failure.

FIGURE 97.4 Midesophageal four chamber TEE view showing severe calcified and fused rheumatic mitral leaflets (arrow).
Diagnosis
The ECG may show left atrial enlargement, right ventricular hypertrophy, and atrial fibrillation. Chest radiography shows straightening of the left heart border, indicating left atrial and pulmonary artery enlargement. Echocardiographic findings include doming of the anterior leaflet, decreased leaflet mobility, increased leaflet calcification and thickness (Fig. 97.4), commissural fusion, calcification of the subvalvular apparatus, increased LA size, and the presence of an LA thrombus (Fig. 97.5). Color flow may show associated mitral regurgitation (MR). Doppler echocardiography allows quantification of severity of MS by calculation of pressure gradients and mitral valve area (Table 97.3; Fig. 97.6). Pressure half-time method is most commonly used to calculate mitral valve area. Evaluation of severity of associated TR and estimation of pulmonary artery systolic pressure can also be done using echocardiography. Cardiac MRI is increasingly being used in the evaluation of stenotic valvular lesions; it measures valve area by planimetry. Its advantage over echocardiography is the lack of dependence upon good echocardiographic windows. Patients with MS often present to the ICU with acute cardiogenic pulmonary edema. Precipitating factors such as infective endocarditis, fever, anxiety, pain, atrial fibrillation, and pregnancy should be identified. Occlusion from an enlarging atrial myxoma should be ruled out. Right-sided heart failure with hepatic dysfunction, acute hemoptysis, systemic embolism, and hoarseness of voice may also be present.

FIGURE 97.5 Prosthetic mitral stenosis with stasis in the left atrium leading to thrombus formation (arrow).
| TABLE 97.3 Echocardiographic Evaluation of Severity of Mitral Stenosis |
 |
Treatment
New-onset atrial fibrillation with hemodynamic instability should be treated with cardioversion. Cardioversion of a patient with atrial fibrillation of unknown duration or that is known to have persisted for more than 48 hours must be preceded by 3 weeks of anticoagulation or by a TEE to exclude the presence of left atrial thrombus. Anticoagulation should be continued for 4 weeks following cardioversion because the enlarged LA remains “stunned” and does not recover a normal contractile state immediately following cardioversion (6). Anticoagulants should also be used in patients with a prior embolic event and left atrial diameter greater than 55 mm by echocardiography (7). Antidysrhythmics, such as amiodarone, may be used to maintain sinus rhythm but should not be expected to provide indefinite success.

FIGURE 97.6 Spectral Doppler through mitral valve showing higher mean gradient and reduced valve area indicating moderate mitral stenosis (compare the values in Table 97.3).
Patients admitted with pulmonary edema benefit from oxygen, diuretics, morphine, anxiolytics, and digoxin; the latter is especially useful in patients with atrial dysrhythmias and congestive heart failure. Sympathetic nervous system activity is increased in patients with MS, worsening the symptom complex; β-blockers are very useful in this situation. Intravenous nesiritide, a synthetic human natriuretic peptide, is also used in the critically ill patients with acute decompensated cardiac failure and pulmonary hypertension (8,9). If the patient is hypotensive, inotropes to improve left ventricular function may not be useful, but may worsen tachycardia and pulmonary edema. Inodilators however may be useful in improving right ventricular dysfunction and reducing pulmonary hypertension. Systemic blood pressure may need to be supported with vasopressors, with the caveat that they may adversely impact pulmonary vascular resistance. The most effective way to support RV function is to decrease pulmonary vascular resistance by administering inhaled vasodilators such as nitric oxide (NO) and epoprostenol. Although inhaled NO decreases PVR and improves right heart function, it may increase left-sided filling pressures in patients with LV dysfunction by increasing blood flow to the pulmonary venous system (10,11). Careful titration and monitoring is required while administering NO to patients with MS as pulmonary edema has been reported in patients with heart failure after treatment with inhaled NO (12). As the LV is usually underfilled, assessment of volume status should focus on the RV. Observation of trends in CVP is useful, especially when a sudden increase occurs, reflecting worsening TR and RV failure.
Emergency invasive intervention is rarely required to relieve MS. If the precipitating events are controlled, elective surgery or balloon valvotomy may be indicated. Balloon valvotomy is possible in patients with pliable leaflets, no commissural fusion, and minimal subvalvular calcification. Because of extensive calcifications and marked anatomic distortion, mitral valve repair may not be possible in rheumatic mitral disease, and valve replacement is necessary in those patients.
Mitral Regurgitation
The mitral valve apparatus is composed of the valve leaflets, mitral annulus, chordae tendineae, and papillary muscles. Any disruption in its integrity may result in MR. One of the major breakthroughs in the management of MR was the functional classification of MR by Carpentier in early 1980s (13) (Table 97.4).
Acute onset of severe MR can lead to acute pulmonary edema and cardiogenic shock. Acute MR may be caused by infection (infective endocarditis with leaflet perforations, vegetations, or periprosthetic valvular leaks); connective tissue or myxomatous disorders (chordal rupture; Fig. 97.7); or ischemic heart disease (infarction and rupture of papillary muscles (Fig. 97.8), or papillary muscle dysfunction due to ischemia). Acute rheumatic mitral valvulitis as the cause of MR is less common today. Patients with MR present to the critical care physician either because of decompensated chronic MR or due to acute onset of severe regurgitation.
| TABLE 97.4 Functional Classification of Mitral Regurgitation (MR) | ||
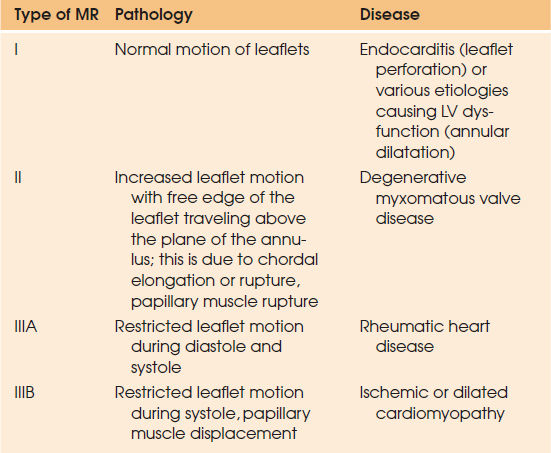 | ||
Pathophysiology
Chronic MR leads to adaptation of the LV by dilatation and eccentric hypertrophy. Over many years of increasing regurgitant volume, systolic function may fail, resulting in decreased LVEF, increased LVEDP, and pulmonary hypertension. Left atrial (LA) dilatation leads to atrial fibrillation. While chronic MR is rarely the direct cause of critical illness, chronic MR often renders the management of a critically ill patient more complex. In acute MR, on the other hand, the regurgitant volume is ejected into the normal, noncompliant left atrium, causing an acute increase in left atrial pressure, which is transmitted to the pulmonary venous system, resulting in pulmonary edema, acute pulmonary hypertension, and RV strain or failure. Sudden volume overload causes further distension of the LV and MV apparatus that increases the regurgitant fraction and decreases forward stroke volume and CO. Systemic vascular resistance rises, further increasing the regurgitant fraction, ultimately leading to cardiogenic shock. The differences in clinical signs between acute and chronic forms of MR are given in Table 97.1.
Diagnosis
The ECG may show atrial fibrillation, left ventricular hypertrophy, right ventricular strain, or myocardial ischemia or infarction. Chest radiograph may show cardiomegaly indicating pre-existing heart disease. Pulmonary venous congestion and/or edema with a normal-sized heart indicate acute MR.
Echocardiography remains the standard for the diagnosis of MR. Bedside TTE often reveals the mechanism of MR. Qualitative and quantitative assessment of MR can be done by color and spectral Doppler methods. TEE may provide better detail because of superior resolution and better definition of anatomic details of the mitral valve apparatus and mechanism and severity of MR. Three-dimensional echocardiography is a revolution in the management of MR by providing accurate diagnosis and precise location of the lesions (Fig. 97.9). Indices of left ventricular function such as LVEF are unreliable in the presence of severe MR. Ejection fraction in MR is increased when contractility is normal. Normal LVEF indicates a significant loss of myocardial function; when LVEF is reduced to 50% or less, advanced myocardial dysfunction is generally present (14). The American Heart Association/American College of Cardiology (AHA/ACC) guidelines recommend medical treatment when EF is less than 30% and surgical treatment, even in asymptomatic patients, with EF less than 60% to prevent progression of disease (7). Urgent coronary angiography is indicated in presence of myocardial ischemia or infarction since the culprit lesion may be amenable to catheter-based or open surgical interventions. In the case of chordal rupture or infective endocarditis without risk factors for CAD, angiography may be deferred.

FIGURE 97.7 Two-dimensional echocardiography showing the posterior leaflet prolapse and ruptured chordae (A) leading to severe anteriorly directed eccentric mitral regurgitation (B).







Full access? Get Clinical Tree


