arterial blood pressure. This hypertensive response, which has a limited clinical relevance, is related to peripheral vasoconstriction. Interestingly, in anaesthetized animals, high intravenous doses of triptans (59,225,323) can decrease blood pressure, probably because of a reduction of sympathetic outflow via an agonist action at central 5-HT1D and/or 5-HT1A receptors (83,148,225). Stimulation of 5-HT1B/1D receptor can also result in vasodilatation mediated by nitric oxide release (139).
 FIGURE 51-1. Chemical structures of sumatriptan and second-generation triptans. |
TABLE 51-1 Affinity and Efficacy of Triptans at Human 5-HT1 Receptorsa | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(see Fig. 51-2) (14,41,184,308,309). The reason for this is not clear, but it may be related to the higher density of 5-HT1B receptors in the cranial compared to coronary arteries (180).
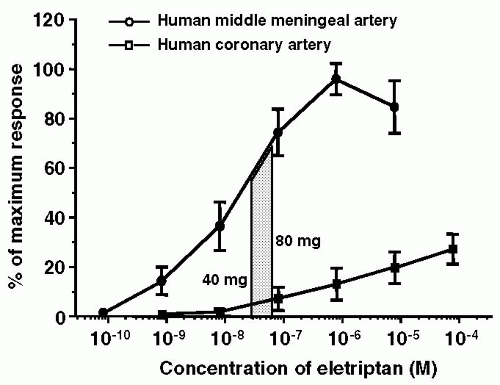 FIGURE 51-2. Concentration-response curves of eletriptan on human middle meningeal and coronary arteries. Superimposed on these curves are the free Cmax (protein-unbound fraction of the maximum plasma concentration) of eletriptan observed in human subjects after oral ingestion of a single 40- or 80-mg tablet. It may be noted that the drug elicits a substantial contraction of the middle meningeal artery, whereas there is only a minimal effect on the coronary artery (184). |
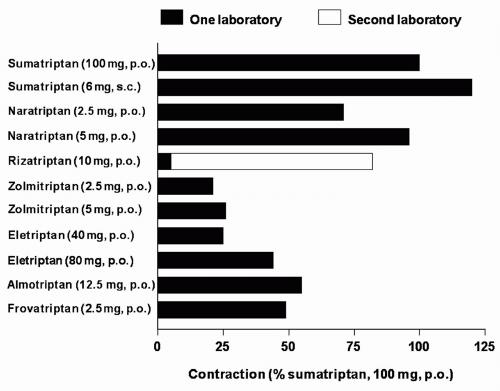 FIGURE 51-3. Predicted contraction of the human isolated coronary artery at therapeutic free Cmax, (protein-unbound fraction of the maximum therapeutic plasma concentration), expressed as percentage of contraction elicited after 100 mg oral sumatriptan (100%). Contraction to sumatriptan (100 mg orally), obtained as a reference, was derived from the same study as the respective compound: subcutaneous sumatriptan, naratriptan, and zolmitriptan (182), rizatriptan (98,178,182), eletriptan (184), almotriptan (14), and frovatriptan (227). Because in the case of rizatriptan studies from two different laboratories were used to predict the coronary artery contraction, values from both laboratories (closed bar) (98,178) and (open bar) (182) are presented. (Redrawn from MaassenVanDenBrink A, Saxena PR. Coronary vasoconstrictor potential of triptans: a review of in vitro pharmacologic data. Headache. 2004;44 [Suppl 1]:S13-S19.) |
several drugs with such an action, including the NK1 receptor antagonist lanepitant (127), the endothelin-receptor antagonist bosentan (198) as well as CP122288 (248), have been found ineffective in migraine. Moreover, the involvement of plasma extravasation itself in migraine has been questioned, as there no changes in retinal permeability during migraine attacks (199). Thus, it is now accepted that the inhibition of dural plasma protein extravasation is not related to the antimigraine action (296). On the other hand, the inhibition of the release of calcitonin gene-related peptide (CGRP) seems to be much more relevant for the antimigraine action, particularly as the novel CGRP receptor antagonist, BIBN4096 (78,161), has been found to be effective in acute migraine (215). Apart from sumatriptan (115) and zolmitriptan (116), donitriptan (174) has been reported to inhibit the release of CGRP during migraine or following electric stimulation of blood vessels or the trigeminal ganglion. However, CGRP release following vanilloid receptor stimulation by capsaicin is not affected by sumatriptan (4).
dorsal horn to block synaptic transmission between axon terminals of the peripheral trigeminovascular neurons and cell bodies of their central counterparts, thus suggesting that the analgesic action of triptans manifests in the absence, but not in the presence, of central sensitization (172).
(140). Based on the pharmacology of triptans (see above), triptans can abort migraine attack by four different mechanisms (Fig. 51-4): (i) a direct contraction of dilated cranial extracerebral blood vessels (62,296), (ii) suppression of neuropeptide (mainly CGRP) release from peripheral nerve endings around blood vessels (118,121), (iii) inhibition of impulse transmission centrally in the trigeminal nucleus caudalis (118,121), and (iv) presynaptic blockade of synaptic transmission between axon terminals of the peripheral trigeminovascular neurons and cell bodies of their central counterparts (172). Although the case for the involvement of neurogenic mechanisms is compelling, in our view, however, the main action of triptans in migraine is the constriction of dilated cranial extracerebral blood vessels via 5-HT1B receptors (97,150,151,244,262). First, the possible contribution of the neuronal effects of triptans mediated via 5-HT1D receptor, which are abundantly present in the trigeminal system (239), has been put in doubt because PNU142633, a selective 5-HT1D receptor agonist, was found ineffective in the treatment of migraine (132). The intrinsic activity of PNU-142633 (70% of 5-HT) at the human 5-HT1D receptor is only a little less than that of sumatriptan (84% of 5-HT), but it is equally effective as sumatriptan and a half-log more potent than sumatriptan in preventing plasma protein extravasation and can reduce increases in cat nucleus trigeminal caudalis blood flow elicited by electrical stimulation of the trigeminal ganglion; importantly, in contrast to sumatriptan it does not reduce carotid blood flow (200). Moreover, besides PNU-142633, there are many other selective 5-HT1D receptor agonists lacking vasoconstrictor activity (179,202), but none has been described as effective in migraine. Another reason that speaks against the involvement of a central action is that sumatriptan (despite a report to the contrary where an extremely high intravenous dose of 3.2 mg/kg was used [159]), does not easily cross the blood-brain barrier (152,162), is comparable in therapeutic effectiveness to centrally penetrative triptans (e.g., rizatriptan and eletriptan); see below. Accordingly, Pascual and Muñoz (229) found no correlation between lipophilicity coefficients and therapeutic efficacy; interestingly there was a significant correlation between lipophilicity and central nervous system (CNS) adverse events. Thus, we submit that the central
mechanisms of triptans may not be necessary for therapeutic efficacy.
TABLE 51-2 Pharmacokinetic Parameters of Triptans in Healthy Volunteers and in Patients with Migrainea | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
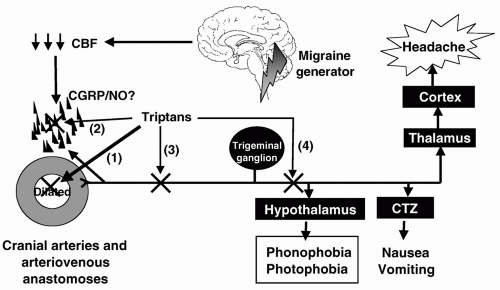 FIGURE 51-4. The pathophysiologic changes in migraine putatively stem from ion leakage through a genetically predisposed faulty channel (217) in the brainstem (321), leading to a decrease in cerebral blood flow, possibly owing to cortical spreading depression (11) and, subsequently, neuropeptide release and dilatation of cranial extracerebral blood vessels (61). The increased pulsation in these blood vessels stimulates the trigeminovascular system, setting in peripheral and central sensitization (172) and leading to headache and associated symptoms (nausea, vomiting, photophobia, and/or phonophobia). The site of action of triptans in aborting migraine attack include: (i) a direct contraction of dilated cranial extracerebral blood vessels, (ii) suppression of neuropeptide (mainly CGRP) release from peripheral nerve endings around blood vessels; (iii) inhibition of impulse transmission centrally in the trigeminal nucleus caudalis; and (iv) presynaptic blockade of synaptic transmission between axon terminals of the peripheral trigeminovascular neurons and cell bodies of their central counterparts. |
have, in most cases, overlapping 95% CIs (Fig. 51-5), and it should be noted that the relative efficacy of two doses or drugs can only be established definitely in comparative trials (Table 51-3).
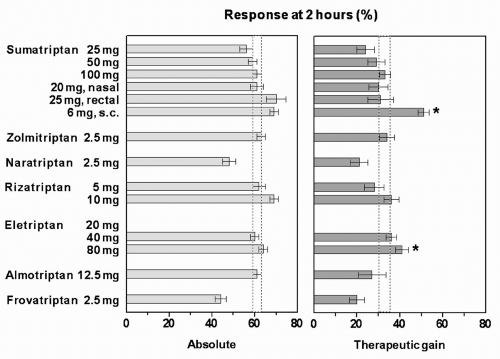 FIGURE 51-5. Mean and 95% confidence intervals (CIs) of absolute and placebo-subtracted (therapeutic gain) headache relief response (i.e., proportion of patients whose moderate or severe headache improves to mild or no pain) at 2 hours after different doses of triptans administered orally, except in case of sumatriptan where nasal, rectal, and subcutaneous routes were also employed (for number of patients treated with each dose of drugs and placebo, see text). The 95% CI of sumatriptan 100 mg (dotted rectangle) has been projected over other responses and significant difference from sumatriptan in the therapeutic gain is indicated by an asterisk (*). (Data collated by P. Tfelt-Hanssen.) |
TABLE 51-3 Randomized Clinical Trials Comparing Triptans | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:







Full access? Get Clinical Tree


