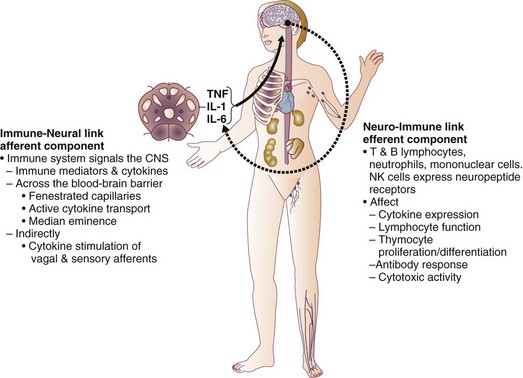
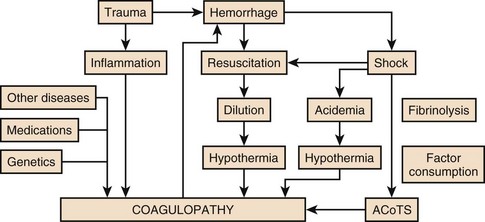
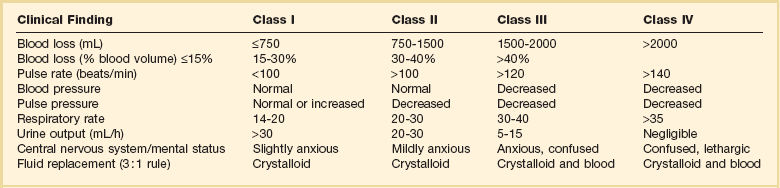
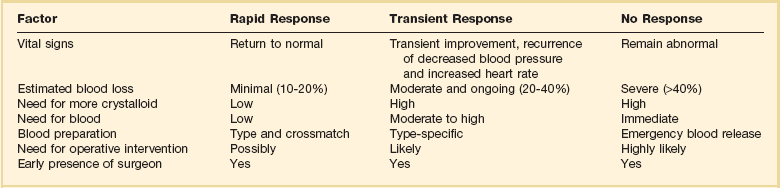
27 In 1934, Blalock suggested four categories of shock: hypovolemic, vasogenic, neurogenic, and cardiogenic.1,2 In more recent clinical practice, additional categories of shock have been proposed.3 Hypovolemic shock, the most common, results from reduction in circulating blood volume. Volume loss may be loss of whole blood, plasma, or extracellular fluid or a combination of all three. Vasogenic shock occurs as a result of changes in the resistance of vessels so that a normal blood volume fails to occupy the available space. Neurogenic shock (spinal shock) is a form of vasogenic shock in which spinal anesthesia or spinal cord injury leads to vasodilation. Septic shock is another form of vasogenic shock in which there is increased capacitance. A decrease in peripheral arterial resistance, a decrease in venous capacitance, and a peripheral arteriovenous maldistribution occur. Cardiogenic shock results from failure of the heart as a pump. Obstructive shock results from mechanical obstruction to cardiac function, as seen with tamponade, tension pneumothorax, or massive pulmonary embolism.4 Traumatic shock includes several components of the conditions mentioned previously.5 Hypovolemia caused by blood loss is compounded by neurogenic, cardiogenic, or obstructive shock plus the vasogenic component of maladaptive mediator cascades initiated by tissue injury. Traumatic shock involves hemorrhage in combination with soft tissue trauma and fractures. As a result, study of pure hemorrhagic shock may have limited relevance to the pathophysiologic condition of traumatic shock. Most studies have shown significant differences in the biologic condition of traumatic shock compared with that of pure hemorrhagic shock based on the activation of mediator cascades.2 Conflicting observations in literature are due at least in part to the assumption that hemorrhagic shock and traumatic shock are identical insults.2,3 Pulmonary complications after simple hemorrhage are uncommon in clinical practice, but pulmonary dysfunction is a common comorbid condition after major trauma with attendant soft tissue or long bone injury.2,6 Activation of mediator systems is far more intense with traumatic shock than with pure hemorrhage.7 Conflicting data regarding changes in cytokine levels after a traumatic insult are likely due to the fact that systemic cytokine levels do not reflect local production of these mediators. Measurement of tissue levels of mediator production may be necessary to determine accurately whether there is upregulation of various mediator systems after trauma or hemorrhage. Soft tissue injury alone upregulates mediator systems.2,8 A small animal study with closed femur fractures showed Kupffer cell activation 30 minutes after injury.9 Another study assessed the effects of skeletal muscle injury in combination with hemorrhage in a porcine model of hemorrhagic shock. To reach a given physiologic end point (reduction in cardiac index and oxygen delivery), hemorrhage of 40% of the blood volume was required in a pure hemorrhagic shock model. If skeletal muscle injury was added, hemorrhage of only 29% of blood volume was necessary to reach the same end point.2,10 The ability to maintain cardiac function after hemorrhage was impaired in this study by superimposition of a soft tissue injury, emphasizing the difference between hemorrhagic shock and traumatic shock. A synergy in activation of neuroendocrine and inflammatory mediator systems is likely when traumatic injury and hemorrhagic shock are present. More recent work describing coagulation changes occurring with injury emphasizes the danger of combined injury and hypoperfusion of soft tissue in failure of appropriate coagulation response.11 The essential homeostatic response to acute blood loss is preservation of cerebral and cardiac perfusion with maintenance of normal blood pressure as sensed by carotid body and aortic arch receptors. Peripheral vasoconstriction and curtailment of fluid excretion are seen. Cardiac contractility and peripheral vascular tone also are altered. Pain, hypoxemia, acidosis, infection, changes in temperature, and availability of substrates such as glucose affect this response. A decrease in blood volume alone without hypotension may activate the hypothalamic-pituitary axis. The magnitude of neuroendocrine response depends not only on the volume of blood loss, but also the rate at which blood loss occurs. This response may be modified by patient age, prescribed medications, preexisting illness, and the use of ethanol or other drugs. With spinal cord transection, operative intervention below the level of injury does not produce typical activation of the hypothalamic-pituitary axis. Similarly, consciousness is unnecessary for activation of this response because it may occur under anesthesia.2,12–16 The initial effect seen with hemorrhage is sympathetic vasoconstriction. Capacitance of the circulatory system is reduced, and aortic arch or carotid sinus baroreceptors respond to changes in blood pressure by modulation of sympathetic tone.2,17 Atrial receptors respond to changes in vascular wall stretch and pressure. Afferent vagal fibers carry signals leading to loss of tonic inhibition of heart rate and immediate activation of thoracolumbar sympathetic outflow with norepinephrine release from postganglionic sympathetic fibers. As blood loss increases, so does the role played by arterial baroreceptors. Another part of this hormonal response is corticotropin-releasing factor secreted by the hypothalamus, vasopressin release, and growth hormone-releasing factor release.12 The clinician sees cool extremities in response to these changes associated with hypovolemia. Venous capacitance also decreases, resulting in accelerated venous return to the heart. Selective arterial vasoconstriction maintains blood flow to the heart and brain until compensation fails. Intense triggering of sympathetic signals is activated when arterial blood pressure decreases to less than 50 mm Hg and is maximally stimulated when systolic blood pressure is less than 15 mm Hg.2 Although metabolic vasoregulation in the heart and brain helps avoid local vasoconstriction, blood flow to other tissues decreases dramatically. Renal blood flow may be reduced to 5% to 10% of normal with acute hypovolemia. Flow to the splanchnic circulation, skin, and skeletal muscle also decreases. These vasoconstrictor responses are mediated by epinephrine and norepinephrine from the adrenal medulla and local sympathetic activity at the vasculature. With increases in acidosis and hydrogen ion concentration, coronary vasodilation occurs as opposed to constriction of arteries in skeletal muscle and the splanchnic circulation.3,18,19 Multiple endocrine responses are seen with trauma and associated hypovolemia. Plasma levels of glucagon, growth hormone, cortisol, and corticotropin (adrenocorticotropic hormone) increase.2,3,5 The renin-angiotensin-aldosterone axis is stimulated with release of vasoconstrictive angiotensin II. Vasopressin release also occurs after hemorrhage, resulting in water absorption in the distal tubule of the kidney. Vasopressin induces splanchnic vasoconstriction. Research suggests that with prolonged hemorrhage, vasopressin depletion may occur, and supplements of this hormone by clinicians may be warranted. Growth hormone and glucagon promote gluconeogenesis, lipolysis, and glycogenolysis. Catecholamines that inhibit insulin release and hyperglycemia and increase blood osmolarity are thought to shift fluid from cells and the interstitium into the intravascular space. More recent data associate hyperglycemia in the setting of injury with adverse outcome, however. The cellular mechanism for this response remains unclear. Loss of fluid or salt through the kidneys also is limited by these hormonal effects, which serve to conserve the circulating blood volume.18,20–22 Compensated acute hypovolemia occurs when the aforementioned mechanisms are sufficient to avoid widespread cellular injury and organ decompensation.2 If volume loss continues, or resuscitation is inadequate, a cycle of decline occurs with regional perfusion defects leading to tissue and microcirculatory changes. Progression from compensated to decompensated and irreversible shock is often defined in retrospect. Frequently, a patient with acute irreversible hemorrhage has been hypotensive for an extended period and cannot be resuscitated despite fluid administration and use of vasoactive drugs.23 Presumed mechanisms in this situation include microcirculatory failure with loss of vasomotor response and integrity of the vascular bed. Patients with subacute but ultimately irreversible shock can be resuscitated initially, but progressive organ injury and end-organ dysfunction follow. In addition to blood loss, extensive research suggests that trauma may be considered an inflammatory disease.24–27 It has been shown that a variety of mediators and indicators of inflammatory response are elevated in severely injured patients. For many of these factors, it could be shown that they were significantly elevated in patients eventually dying compared with survivors, and that prediction of outcome is possible with a significant degree of accuracy. Peak inflammatory activity as measured by plasma values has been noted within hours of injury. Although it cannot at present be decided which of these parameters may play a direct pathophysiologic role in development and promotion of inflammatory response and consecutive organ dysfunction, and which is an indicator of this reaction, inflammatory mediators may reflect pathophysiologically relevant disturbances set off by tissue injury and blood loss with consecutive ischemia and reperfusion incidents.28 Shock after trauma differs from pure hypovolemic shock in that effects of release of mediators by tissue injury are superimposed on hypovolemia. It also is clear that not all damage after shock is the result of tissue hypoxia, and that much of cellular damage follows reperfusion and subsequent inflammation. Loci of this inflammatory response are the wound, with activation of macrophages and production of proinflammatory mediators, and the microcirculation, with activation of blood elements and the endothelium.28,29 With blood loss, classic circulatory variables, such as systolic blood pressure, remain normal or supranormal until 30% of blood loss occurs.2,30 With progressive cellular hypoxia, mitochondria still may be able to metabolize oxygen.2 Nonetheless, with significant hypovolemia, total oxygen available to tissue is severely reduced, causing anaerobic metabolism, which is energy inefficient because one molecule of glucose is no longer able to contribute to resynthesis of 32 mol of adenosine triphosphate but only to 2 mol. Glucose must reach cells through the circulation, which is critically reduced. In addition, the end product is no longer carbon dioxide, which can be eliminated by ventilation, but lactic acid and hydrogen ions, leading to metabolic acidosis. Acidosis drives cellular swelling with loss of extracellular fluid volume into the cells. Lactate finally is metabolized by the liver, which also is hypoxic. Transcapillary refill and lymph flow direct interstitial fluid to increase the circulating blood volume, but ultimately capillaries are damaged by hypoxia and the action of activated neutrophils, which increases interstitial edema. Finally, autoregulation of microcirculation is destroyed, leading to fluid sequestration and sludging in the microvasculature. These factors are responsible for increased diffusion distance for oxygen from capillaries to the mitochondria, which further impairs oxygen extraction. Tissue hypoxia also is the most potent stimulus for proinflammatory activation of macrophages and release of vasoactive or arachidonic acid metabolites, such as prostaglandins and thromboxane. Hypovolemia, shock, and any other cause of brain hypoxia also are detrimental to recovery, particularly in patients with head injury because these conditions induce secondary brain damage. Although a variety of initiating events may occur, the subsequent inflammatory response is qualitatively similar.2 Local activation of the complement cascade produces anaphylatoxins, which are strong attractants and stimulants of neutrophils. Local endothelium expresses endothelial leukocyte adhesion molecules, which attract the neutrophil population. Activated neutrophils also express adhesion molecules, leading to aggregation, margination in the vascular endothelium, and migration through vessel walls at the area of injury. This inflammatory response produces a respiratory burst with formation of oxygen radicals and synthesis of proteolytic enzymes (elastase). Local release of bradykinin, histamine, and prostaglandin induces local vasodilation and increased capillary permeability from macromolecules, resulting in a protein-rich exudate. Local phagocytes release messenger molecules, such as granulocyte-macrophage colony-stimulating factor and macrophage colony-stimulating factor, which activate the bone marrow to produce more inflammatory cells. Neutrophils injure otherwise healthy tissues.2,31–34 In a slower response, the monocyte population is attracted to the site of injury, where it differentiates to macrophages and contributes to the inflammatory process by phagocytosing and killing bacteria or disposing of necrotic tissue or both. Macrophages are activated further by triggers such as hypoxia or C5a, macrophage-activating factor, and interleukin (IL)-1-like activity from neutrophils. On stimulation, macrophages release a variety of classes of secretory products, which may be proinflammatory (proteolytic enzymes, oxygen radicals, IL-1, IL-6, tumor necrosis factor) or anti-inflammatory (IL-10, prostaglandin E2). Macrophage mediators such as prostaglandin E2, tumor necrosis factor, IL-1, IL-2, and IL-6 provide systemic signals adapting metabolic and defense mechanisms. Macrophages take several days after activation to develop full inflammatory capacity. They also may release nitric oxide and cytotoxic radicals. In the setting of injury, this local inflammatory process spills over to cause an exaggerated systemic response with inflammatory damage to otherwise healthy cells and organs distant to the site of injury. Secondary infection may occur in the compromised host, leading to generalized inflammation and multiorgan dysfunction (Box 27.1).2,35,36 More recent work examines the link between the autonomic nervous system and modulation of immune response during traumatic injury. Anatomic interactions with immune-competent cells have been identified, and functional consequences of this interaction in the host are now being examined. Integrated hemodynamic, metabolic, behavioral, and immune responses allowing host adaptation are the stress response.37–41 Catecholamines are neurotransmitters that affect immune response humorally through circulating adrenal-derived epinephrine and locally through neuronal release of norepinephrine. There is anatomic evidence of central nervous system (CNS)–lymphoid organ connection through autonomic and sensory fibers and immune tissues, including bone marrow, thymus, spleen, and lymph nodes.37 This sympathetic innervation of lymphoid organs is found across species and has been confirmed by immunohistochemistry. In bone marrow, myelinated and nonmyelinated fibers are distributed with vascular plexuses where they influence hematopoiesis and cell migration. In the lungs, noradrenergic nerve fibers supply tracheobronchial smooth muscle and glands. In addition, nerve fibers have been shown throughout the different compartments of the bronchus-associated lymphoid tissue forming close contact with mast cells, cells of the macrophage/monocyte lineage, or other lymph node cells. In the thymus, noradrenergic nerve fibers have been localized in the subcapsular, cortical, and corticomedullary regions associated with blood vessels and intralobular septa branching into cortical parenchyma where they reach to thymocytes.37,42 The functional effects of catecholamines on cells of the immune system have been confirmed in human volunteers. In addition, relevance of this control mechanism and the implications for dysregulation have been shown by rapid systemic release of IL-10 and the high incidence of infection in patients with sympathetic storm from accidental or iatrogenic brain trauma.37 Although detrimental effects of sustained and exaggerated sympathetic nervous system activation on cardiovascular and metabolic homeostasis have long been recognized, attention is now directed to the likelihood of immune dysregulation as well. The neuroimmune axis is a bidirectional network composed of descending pathways linking the CNS to peripheral immune tissues and a parallel afferent arm linking the immune system with the CNS. The integrity of this loop allows for communication between the CNS and peripheral immune system integrating neuronal and immune signals in the periphery and in the CNS. Cells from the immune system express functional receptors and signal transduction pathway components for several neuroendocrine mediators allowing functional cellular responses to agonist stimulation. Similarly, cells in the CNS are capable of synthesizing, secreting, and responding to inflammatory and immune molecules. There is considerable evidence that the peripheral immune system can signal the brain to elicit a sickness response during infection, inflammation, and injury. Peripheral immune molecules such as cytokines influence CNS action through mechanisms including entry into the brain through a saturable transport mechanism or through areas that lack the blood-brain barrier. Afferent neurons of the vagus nerve also are activated (Fig. 27.1).43–45 Severe trauma is characterized by the classic activation of the sympathetic nervous system and the recently recognized contribution of the inflammatory and neuroimmune response to injury.37 The sympathetic nervous system has significant anatomic and functional interaction with cells of the immune system and plays an important role in control of the magnitude of early inflammatory response to injury by ensuring expression of adequate cytokine balance.37 Sympathetic neural pathways exert direct effects on cells of the immune system, affecting cytokine expression, lymphocyte function, and cytotoxic activity. In return, the inflammatory mediators released communicate with the CNS through stimulation of sensory and vagal afferents or by crossing the blood-brain barrier through active transport mechanisms and pathways allowing access to hypothalamic-pituitary structures. Immune-derived mediators, such as cytokines and chemokines, can modulate neurotransmission affecting activation of descending autonomic and neuroendocrine pathways.37 Hemorrhagic shock accounts for a significant number of deaths in patients arriving at hospital with acute injury. Patients with uncontrolled hemorrhage continue to die despite adoption of new surgical techniques with improved transport and emergency care.46,47 Coagulopathy, occurring even before resuscitation, contributes significantly to the morbidity associated with bleeding.48,49 Recognition of the morbidity associated with bleeding and coagulation abnormality dates to the Vietnam conflict. At that time, standard tests including prothrombin time (PT) and partial thromboplastin time (PTT) correlated poorly with effectiveness of acute resuscitation efforts. Similar work in the late 1970s was performed in civilian patients receiving massive transfusion. Again, PT, PTT, and bleeding time were only helpful if markedly prolonged.50,51 Studies in the 1970s and 1980s provided additional detail regarding the limitation of simple laboratory parameters and factor levels.51,52 In a study of multiple patients requiring massive transfusion, platelet counts fell in proportion to the size of transfusion although factors V and VIII correlated poorly with the volume of blood transfused. Where coagulopathy appeared, patients seemed to respond to platelet administration. In subsequent studies, patients receiving a large number of blood products were followed for microvascular bleeding. Moderate deficiencies in clotting factors were common, but they were not associated with microvascular bleeding. Microvascular bleeding was associated with severe coagulation abnormalities such as clotting factor levels less than 20% of control values. In statistical analysis, clotting factor activities less than 20% of control levels were predicted by significant prolongation of PT and PTT. These earlier investigators also suggested that empiric blood replacement formulas available at the time were not likely to prevent microvascular bleeding because consumption of platelets or clotting factors did not consistently appear and simple dilution caused by resuscitation fluids frequently did not correspond to microvascular bleeding.52 The attention of the American trauma community was drawn to coagulopathy after trauma with the description of the “bloody vicious cycle” by the Denver health team over 20 years ago.48 These investigators noted the contribution of hypothermia, acidosis, and hemodilution associated with inadequate resuscitation and excessive use of crystalloids. Subsequent work extended these observations describing early coagulopathy that could be independent of clotting factor deficiency.53 In a more recent trial, early coagulopathy was noted in the setting of severe injury, which was present in the field, prior to emergency department arrival and initiation of fluid resuscitation. Coagulopathic patients were at increased risk for organ failure and death. In a study questioning historical transfusion practice emphasizing administration of packed red blood cells (PRBCs) in the setting of massive trauma, Hirshberg and coworkers, using clinical data, developed a computer model designed to capture interactions between bleeding, hemodynamics, hemodilution, and blood component replacement during severe hemorrhage. Resuscitation options were offered in this model and their effectiveness evaluated.54 After setting thresholds for acceptable loss of clotting factors, platelets, and fibrinogen, the authors modeled behavior of coagulation during rapid exsanguation without clotting factor or platelet replacement. The PT reached a critical level first followed by fibrinogen and platelets. If patients were resuscitated with small amounts of crystalloid, leaving overall blood volume reduced, the effective life of components in the coagulation cascade was increased. More aggressive fresh frozen plasma (FFP) replacement in the patient with significant bleeding was supported by this model. The optimal ratio for administration of FFP to PRBCs in this analysis was 2 : 3. Delayed administration of FFP led to critical clotting factor deficiency regardless of subsequent administration of FFP. Fibrinogen depletion was easier to correct. After administration of 5 units of PRBCs, the hemostatic threshold for fibrinogen was not exceeded if a FFP-to-PRBC ratio of 4 : 5 was employed. Analysis of platelet dilution demonstrated that even if platelet replacement was delayed until 10 units of PRBCs were infused, critical platelet dilution was prevented with a subsequent platelet-to-PRBC ratio of 8 : 10.54 Brohi and coworkers from the United Kingdom helped to reinvigorate discussion of coagulopathy after injury by adding new coagulation laboratory techniques to previous clinical observations.55 After reviewing over 1000 cases, patients with acute coagulopathy after injury had higher mortality rates throughout the spectrum of Injury Severity Scores (ISS). Contrary to historical teaching that coagulopathy was a function of hemodilution with massive crystalloid resuscitation, these authors noted that the incidence of coagulopathy increased with severity of injury but not necessarily in relationship to the volume of intravenous fluid administered to patients. Brohi and others helped to reemphasize the observation that acute coagulopathy could occur before significant fluid administration, which was attributable to the injury itself and proportional to the volume of injured tissue. Development of coagulopathy was an independent predictor of poor outcome. Mediators associated with tissue trauma including humoral and cellular immune system activation with coagulation, fibrinolysis, complement, and kallikrein cascades have been associated with changes in hemostatic mechanisms similar to those identified in the setting of sepsis.55–57 Factors contributing to coagulopathy in the setting of injury have been further reviewed.58 Hypothermia relates to development of coagulopathy by reduction in platelet aggregation and decreased function of coagulation factors in nondiluted blood. Patients with temperature reduction below 34° C had elevated PT and PTT. Coagulation, like most biologic enzyme systems, works best at normal temperature. Similarly, acidosis occurring in the setting of trauma as a result of bleeding and hypotension also contributes to clotting failure. Animal work shows that a pH less than 7.20 is associated with hemostatic impairment. Platelet dysfunction and coagulation enzyme system changes are noted when blood from healthy volunteers is subjected to an acidic environment.59,60 Hess and coworkers, as part of an international medical collaboration, developed a literature review to increase awareness of coagulopathy independent of crystalloid administration following trauma.57 The key initiating factor is volume of tissue injury. Patients with severe tissue injury but no physiologic derangement, however, rarely present with coagulopathy and have a lower mortality rate.61,62 Tissue damage initiates coagulation as endothelial injury at the site of trauma leads to exposure of subendothelial collagen and activation of the coagulation cascade. Hyperfibrinolysis is seen as a direct consequence of the combination of tissue injury and shock. Endothelial injury accelerates fibrinolysis because of direct release of tissue plasminogen activator.57,63 Tissue plasminogen activator expression by endothelium is increased in the presence of thrombin. Fibrinolysis is accelerated because of the combined effects of endothelial tissue plasminogen activator release with ischemia and inhibition of plasminogen activator inhibitor in shock. Although hyperfibrinolysis may focus clot propagation on sites of actual vascular injury, with widespread insults, this localization may be lost. A number of important cofactors must be present to stimulate coagulopathy in the setting of injury.57 Shock is a dose-dependent cause of tissue hypoperfusion. Elevated base deficit has been associated with coagulopathy in as many as 25% of patients in one large study. Progression of shock appears to result in hyperfibrinolysis. One mediator implicated in coagulopathy after injury is activated protein C. Immediate postinjury coagulopathy is likely a combination of effects caused by large volume tissue trauma and hypoperfusion (Fig. 27.2).57 As will be discussed later, equivalent ratios of FFP, PRBCs, and platelets are now considered for management of significant hemorrhage with coagulopathy after injury. Hypothermia and acidemia must be controlled to reduce their impact on enzyme systems.64 Similar to sepsis, cross-talk has been noted between coagulation and inflammation systems with injury. Activation of coagulation proteases may induce inappropriate inflammation with activation of cascades such as complement and platelet degranulation.65,66 Trauma patients are initially coagulopathic with increased bleeding. This condition may progress to a hypercoagulable state, putting them at risk for thrombotic events. This late thrombotic state bears similarities with coagulopathy of severe sepsis and depletion of protein C. Injured and septic patients share a propensity toward multiple organ failure and prothrombotic states.67,68 Warmed isotonic electrolyte solutions are recommended for initial resuscitation of traumatic shock by the Committee on Trauma of the American College of Surgeons. This type of fluid provides transient intravascular expansion and stabilizes the intravascular volume by replacing accompanying fluid losses into the interstitial and intracellular spaces. Lactated Ringer’s solution is the initial fluid of choice. Normal saline is the second choice. Normal saline has the potential to cause hyperchloremic acidosis. This complication is more likely if renal function is compromised (Table 27.1).69 Table 27.1 Estimated Fluid and Blood Losses Based on Initial Clinical Presentation* *This is the standard approach to resuscitation of shock after injury as described in the Advanced Trauma Life Support course promulgated by the Committee on Trauma of the American College of Surgeons. The crystalloid of choice used in resuscitation is lactated Ringer’s solution. Clinical parameters are used to estimate the degree of blood loss, and fluid resuscitation begins with 1-2 L of lactated Ringer’s solution given through large-bore peripheral intravenous lines. When the response to resuscitation is limited or transient, O-negative or type-specific blood is added to resuscitation while the cause of shock is sought and additional treatment is given. From American College of Surgeons Committee on Trauma: Advanced Trauma Life Support for Doctors, 7th ed. Chicago, American College of Surgeons, 2004, pp 69-85. An initial warm fluid bolus is given rapidly—usually 1 to 2 L for an adult and 20 mL/kg for a child.45 Patient response is observed during this initial fluid resuscitation, and subsequent therapeutic decisions are based on this response. The required amount of fluid and blood is difficult to predict on initial evaluation of the patient. A rough guideline promulgated by the American College of Surgeons for the total amount of crystalloid volume acutely required is 3 mL of crystalloid fluid to replace each 1 mL of blood loss, allowing for restitution of plasma volume lost into interstitial and intracellular spaces. It is most important, however, to assess patient response to fluid resuscitation and evidence of adequate end-organ perfusion as measured by urine output and level of consciousness, rather than provide fluid based on a specific formula. If the amount of fluid required to restore or maintain adequate end-organ function exceeds the previously mentioned estimates, careful reassessment of the situation and exploration for unrecognized injuries, bleeding, or other causes of shock are necessary (Table 27.2). Table 27.2 Responses to Initial Fluid Resuscitation* *The Advanced Trauma Life Support course advocates ongoing evaluation of patient response to initial fluid administration. Patients with no response frequently require emergent blood transfusion and transfer to the operating room. Patients with transient response also frequently require operative intervention. Most patients, particularly in centers seeing blunt injury, respond rapidly to an initial 1-2 L of crystalloid and are cleared to proceed to more detailed imaging to determine internal injuries after normalization of clinical parameters. From American College of Surgeons Committee on Trauma: Advanced Trauma Life Support for Doctors, 7th ed. Chicago, American College of Surgeons, 2004, pp 69-85. In clinical practice, large volume resuscitation with lactated Ringer’s solution has become common in trauma care.70 However, recent military and laboratory work features a growing concern about tissue edema from large volume resuscitation. In recent decades, a persisting picture of acute lung injury due to increased filtration across pulmonary microcapillaries with pulmonary inflammation emerged. This process would later be called the acute respiratory distress syndrome.71 Other observations included increased interstitial fluid of gut and heart tissues, abdominal compartment syndrome, extremity compartment syndrome in uninjured extremities, and pericardial effusion.72,73 Hemorrhage is a multifactorial disease; circulatory and inflammatory effects of hemorrhagic shock occur simultaneously. Unfortunately, laboratory studies have repeatedly shown that the choice of resuscitation fluid may worsen hemorrhage-induced cellular dysfunction, immune modulation, and inflammation. Fluids affect neutrophil activity by changing life span, activation, and gene expression. Resuscitation fluids also enhance inflammatory cascade through upregulation of cellular receptors and proinflammatory mediators. The choice of fluid also affects cellular gene expression, apoptotic cell death, and extracellular matrix integrity.70,74–76 Of isotonic crystalloids, lactated Ringer’s solution has been most extensively studied to determine its role in hemorrhage-induced immune dysfunction, inflammation, and management of ischemia and reperfusion injury. Lactated Ringer’s solution has been shown to upgrade vascular endothelial adhesion molecules and to increase expression of CD11b and CD18 binding sites on neutrophils. Neutrophil oxidative burst is also stimulated by lactated Ringer’s solution. In other organs, Ringer’s lactate has been found to increase apoptosis in the bowel, the liver, and the lung with multiple cell types affected including macrophages, endothelial cells, epithelial cells, and smooth muscle cells.70,77 Despite laboratory findings about the dangers of lactated Ringer’s solution, it remains the fluid of choice in many centers and the recommended fluid of the Advanced Trauma Life Support (ATLS) protocol. Efforts have been made to examine why lactated Ringer’s solution is cytotoxic and identify ways to improve it. Traditionally, lactated Ringer’s solution came in racemic form; laboratory work implicates the D-isomer of lactate as its primary toxic component.78 The D-isomer was found to increase neutrophil oxidative burst, enhance apoptosis, and drive inflammation. The L-isomer of lactate may confer immune protection through attenuation of neutrophil activation, alteration of leukocyte gene expression, and reduction in apoptosis.79,80 Hyperoncotic colloid solutions have also been studied in resuscitation roles for traumatic hemorrhage. The natural colloid albumin does not induce neutrophil oxidative burst and may confer a protective immunologic effect by decreasing neutrophil expression of adhesion molecules.81 At present, albumin sees little application in resuscitation at the scene of injury but has been investigated in critical care practice. An artificial colloid, 6% hetastarch, has been found to have a number of deleterious resuscitation effects in animal models including increased neutrophil oxidative burst and pulmonary apoptosis. Beneficial effects include decreased neutrophil migration. At present, natural and artificial colloids have failed to show clinical benefits in comparison with crystalloid solutions.82,83 Laboratory concerns and lack of a positive clinical outcomes mandate argue against the use of colloids in early resuscitation of hemorrhagic shock. Recent reviews suggest important differences in safety among colloids. Examination of data comparing colloids with crystalloids must take into account materials employed. When albumin was used as a reference, the incidence ratio for anaphylactoid reactions was 4.51 after administration of hydroxyethyl starch, 2.32 after dextran, and 12.4 after gelatin. Artificial colloid administration was consistently associated with coagulopathy and clinical bleeding, most frequently in cardiac surgery patients receiving starches. Albumin had the lowest rate of total adverse events and serious adverse events.84 Although albumin is isolated from human plasma, no evidence of viral disease transmission has been consistently identified. Life-threatening anaphylactoid reactions were infrequent for all colloids. Hydroxyethyl starch, as compared with albumin, more than quadrupled the incidence of anaphylactic reactions, whereas dextran more than doubled them. The incidence of these reactions in recipients of gelatin was greater by an order of magnitude than after albumin infusion. Because artificial colloids are derived from nonhuman source materials, they may be recognized as foreign and are more likely to provoke this immune-mediated response. The foreign nature of artificial colloids also may hinder metabolic clearance and promote tissue deposition. On the basis of extensive evidence, albumin is the safest colloid for consideration in resuscitation of traumatic shock.84 Although factors such as desirability of anticoagulant activity may favor other artificial colloids, this is not true in the setting of injury.85–87 Multicenter data comparing albumin and saline for fluid resuscitation were obtained in Australia and published in 2004.88 Nearly 7000 patients were randomly assigned to administration of 4% albumin or normal saline for intravascular fluid resuscitation procedures. Mortality rate and the incidence of single and multiple organ dysfunction were comparable in the two groups. Subset analysis suggests, however, poorer outcomes in the setting of injury. In the subgroup of 140 patients included with principal diagnoses of trauma, a treatment effect seemed to favor administration of saline. In this trial, the increased relative risk of death among patients with trauma compared with patients without trauma resulted from an excess number of deaths among patients who had trauma with brain injury. The difference in mortality rates between albumin and saline groups among patients with trauma involving brain injury must be viewed cautiously because the number of involved subjects is small. In the Australian trial, patients with traumatic brain injury constituted only 7% of the study population, and the excess number of deaths in the albumin group was 21. Other parameters that could be helpful in evaluation of the impact of albumin in the setting of brain injury, such as functional neurologic status, were not provided. In contrast with the experience in trauma, the Australian trial suggests some evidence of treatment benefit favoring administration of albumin in patients with severe sepsis. Given contemporary resuscitation technology, factors influencing the choice of resuscitation for critically ill patients include specific clinician concerns, treatment tolerance, safety, and cost. Increased transmembrane sodium gradient caused by hypertonic saline generates intravascular volume expansion similar to hyperoncotic colloids and superior to conventional isotonic crystalloids such as lactated Ringer’s solution and normal saline. Animal models suggest that hypertonic saline solutions dilate precapillary arterioles and shunt oxygen to vital organs.89,90 Hypertonic saline solutions also have fewer proinflammatory properties than other clinical crystalloids and colloids. Hypertonic saline does not induce expression of inflammatory cytokine receptor genes in multiple studies and blunts hemorrhage-induced increase in plasma levels of proinflammatory cytokines, IL-10, and granulocyte-macrophage colony-stimulating factor. Hypertonic saline also does not increase apoptotic cell death in liver, lung, or bowel.70 What about the impact of hypertonic saline and associated hypernatremia on head injury?91 Studies in experimental animals and humans suggest that hypertonic saline may be highly effective in treating head injury, either alone or associated with hemorrhagic hypotension. Tissue swelling in a closed cranium threatens to cause major pressure-induced brain damage or death, and concomitant hemorrhage hypotension reduces cerebral oxygen delivery, resulting in a secondary ischemic insult. Historical data suggest a twofold higher incidence of adverse outcomes in patients with brain injury combined with hypotension. Early data suggest that patients treated with hypertonic saline with dextran are more likely to survive to discharge than individuals treated with standard resuscitation care.92,93 Mixture of hypertonic saline with dextran has been the most extensively tested hypertonic-hyperoncotic fluid.70 Use of combinations of hypertonic saline and dextran suggests that this material is effective in expanding plasma volume, restoring hemodynamics, and improving microcirculatory perfusion. In the laboratory, hypertonic saline and dextran solutions blunt hemorrhage-induced inflammatory response by neutrophils and, in clinical trials, decreased adhesion molecule expression.94 As with hypertonic saline solutions, there has been concern that hypertonic saline mixed with dextran could accelerate hemorrhage, increase mortality rate, and cause hypernatremia and hyperchloremia.95 Despite multiple clinical trials comparing hypertonic saline and dextran solutions to more traditional resuscitation products, no improvement in mortality rate or change in the pattern of organ failure is seen.96 Mechanisms by which hypertonic/hyperoncotic resuscitation may be effective in models of head injury and hemorrhage show reduction in water content in noninjured portions of the brain with reduction in intracranial pressure and cerebral edema. In a large animal model, when hypertonic saline was compared with a synthetic colloid, colloid alone had no effect on brain water content.97,98 Plasma and blood were the fluid replacements of choice in traumatic shock until the early 1960s, when a variety of investigators showed the need to replace the extracellular fluid deficit with crystalloid solutions. These observations were followed by a variety of clinical studies comparing colloid, typically albumin, solutions with crystalloids, typically lactated Ringer’s solution. Consistent with early studies, colloids, when given on an equal volume basis, more effectively increase cardiac output and oxygen transport. Another finding of this early work was the need to give crystalloids in far greater quantities than colloids to achieve consistent hemodynamic objectives.99,100 Later studies from the Vietnam era compared resuscitation of patients who were given whole blood and crystalloids with patients given whole blood plus 5% albumin. Fluid infusion volumes were far higher in the patients given crystalloid solutions. There was no evidence of pulmonary edema, and patients treated with crystalloids seemed to fare better than patients treated with resuscitation containing albumin. Albumin seemed to have less effect on restoration of renal function with suggestion of detrimental effects in pulmonary response, myocardial contractility, and coagulation. Large animal models suggested that pulmonary compromise could relate to increased capillary permeability to albumin. Increased losses of albumin to the heart, kidneys, liver, and brain also were reported.99,101 More extensive studies in injured patients supported reservations regarding the use of albumin. Evaluation of patients randomly selected to receive 150 g of albumin per day intraoperatively and postoperatively noted poorer outcomes than in patients receiving lactated Ringer’s solution. Both groups received whole blood and FFP. Patients treated with albumin required greater ventilator support and had poorer oxygenation.99,102,103 In another carefully conducted trial of patients with multiple trauma, no differences in cardiopulmonary function between patients resuscitated with lactated Ringer’s solution and patients given 5% albumin and lactated Ringer’s solution were identified.104 Normal cardiac index was used as a therapeutic end point. To maintain adequate cardiac output, patients who received crystalloids required far more resuscitation volume than patients treated with albumin. These authors concluded that cardiac output was an appropriate end point for resuscitation, and that no advantage was accrued based on the type of fluid employed. A clear cost advantage of crystalloids was identified.99 Guyton and Lindsey105 examined the effect of colloid oncotic pressure on pulmonary edema. They observed that reducing the serum protein level lowered the threshold of left atrial pressure at which pulmonary edema could occur. Zarins and colleagues106 subsequently showed that a low colloid oncotic pressure alone did not cause an elevation in extravascular lung water. Because of the remarkable efficiency of pulmonary lymphatics, arterial blood gases, shunt fraction, and lung compliance were unchanged despite a 14% increase in body weight caused by infusion of lactated Ringer’s solution to keep high pulmonary artery occlusion pressures. No pulmonary edema was created despite the presence of ascites and marked peripheral edema. Demling and coworkers107 confirmed these findings with a chronic lung lymph fistula in sheep. Holcroft and coworkers108 produced pulmonary edema in baboons during resuscitation from hemorrhage by continuously administering large volumes of lactated Ringer’s solution sufficient to elevate pulmonary artery occlusion pressures 15 mm Hg above baseline levels. With cessation of infusion, filling pressure rapidly returned to normal. Despite work from multiple groups suggesting that simple replacement of PRBCs was not a sufficient answer for the most severely injured patient, particularly in the setting of coagulopathy, the concept of combination blood component replacement remained outside the mainstream of trauma care for over 20 years.48,52,109 It took armed conflicts and experience in a multinational group of trauma centers to bring awareness of the need for multiple blood component therapy in massive bleeding to the level of general trauma practice. The 1970s and 1980s saw several groups propose resuscitation of significant hemorrhage with combinations of blood components. Kashuk and Moore proposed multicomponent blood therapy in patients with significant vascular injury.48 In a study of patients with major abdominal vascular injury, Kashuk and coworkers noted frequent deviation from a standard ratio of 4 : 1 or 5 : 1 for units of PRBCs to units of FFP. The ratio was 8 : 1 in nonsurvivors and 9 : 1 where overt coagulopathy was noted. Fifty-one percent of patients in this series were coagulopathic after vascular control was obtained. Using multivariate analysis, Ciavarella and coworkers from the Puget Sound Blood Center and Harborview Medical Center proposed aggressive supplementation of platelets in the setting of massive transfusion. These investigators noted that platelet counts below 50 × 109/L correlated highly with microvascular bleeding in trauma and surgery patients. Fibrinogen repletion was also emphasized. Guides to resuscitation included fibrinogen level, PT, and PTT. Supplemental FFP or cryoprecipitate was recommended for low fibrinogen levels.52 Lucas and Ledgerwood, summarizing extensive preclinical and clinical studies, suggested administration of FFP after 6 units of PRBCs had been infused. Additional FFP was recommended for every five additional PRBC transfusions. Monitoring included platelet count, PT, and PTT after each 5 units of PRBCs are administered. Platelet transfusion is generally unnecessary unless the platelet count falls below 50,000.109 Rhee and coworkers, using the massive database of the Los Angeles County Level I Trauma Center, examined transfusion practices in 25,000 patients.110 Approximately 16% of these patients received a blood transfusion. Massive transfusion (≥10 units of PRBCs per day) occurred in 11.4% of transfused patients. After excluding head-injured patients, these authors studied approximately 400 individuals. A trend toward increasing FFP use was noted during the 6 years of data that were reviewed (January 2000 to December 2005). Logistic regression identified the ratio of FFP to PRBC use as an independent predictor of survival. With a higher ratio of FFP : PRBC, a greater probability of survival was noted. The optimal ratio in this analysis was an FFP : PRBC ratio of 1 : 3 or less. Rhee and coworkers provide a large retrospective data set demonstrating that earlier more aggressive plasma replacement can be associated with improved outcomes after bleeding requiring massive transfusion. Ratios derived in this massive retrospective data review support the observations of Hirshberg and coworkers.54 Like the data presented by Kashuk and coworkers in another widely cited report, this retrospective data set suggests improved clinical outcome with increased administration of FFP.111 Another view of damage control hematology comes from Vanderbilt University Medical Center in Nashville, Tennessee. This group implemented a trauma exsanguination protocol involving acute administration of 10 units PRBC with 4 units FFP and 2 units platelets. In an 18-month period, 90 patients received this resuscitation and were compared to a historical set of control subjects. The group of patients receiving the trauma exsanguination protocol as described by these investigators had lower mortality rates, higher blood product use in initial operative procedures, and more frequent use of products in the initial 24 hours, though overall blood product consumption during hospitalization was decreased.112 The strongest multicenter civilian data report examining the impact of plasma and platelet administration along with red blood cells on outcome in massive transfusion comes from Holcomb and coworkers.113 These investigators report over 450 patients obtained from 16 adult and pediatric centers. Overall survival in this group is 59%. Patients were gravely ill as reflected by an admission base deficit of −11.7, pH 7.2, Glasgow Coma Scale score of 9, and a mean ISS of 32. Examination of multicenter data reflects an improvement in outcome as the ratio of FFP to PRBCs administered approaches 1. FFP, however, is not the sole solution to improved coagulation response in acute injury. These workers also examined the relationship of aggressive plasma and platelet administration in these patients. Optimal outcome in this massive transfusion group was obtained with aggressive platelet as well as plasma administration. Worst outcomes were seen when aggressive administration of plasma and platelets did not take place. When either FFP or platelets were given in higher proportion in relationship to PRBCs intermediate results were obtained. Not surprisingly, the cause of death that was favorably affected was truncal hemorrhage. A summary statement comes from Holcomb and a combination of military and civilian investigators.56,57
Traumatic Shock and Tissue Hypoperfusion
Nonsurgical Management
Classic Neuroendocrine Response
Inflammation in Shock after Injury
Cellular Energetics
Immune Mediator Cascades
Neuroimmune Response to Trauma
Acute Coagulopathy after Trauma
Historical Perspective
Recent Studies
Fluid Therapy
Isotonic Crystalloids
Colloids
Hypertonic Saline
Hypertonic-Hyperoncotic Fluids
Crystalloids Versus Colloids
Blood Component Therapy

Full access? Get Clinical Tree


Anesthesia Key
Fastest Anesthesia & Intensive Care & Emergency Medicine Insight Engine