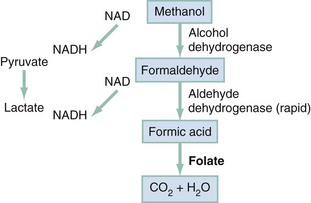
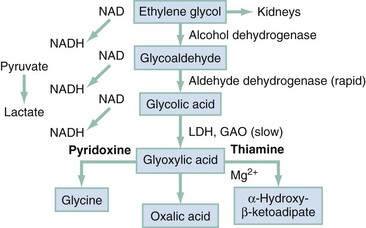
Chapter 155 Methanol is absorbed rapidly from the gastrointestinal tract, and blood levels peak 30 to 60 minutes after ingestion.1 Transdermal and respiratory tract absorption also has resulted in toxicity, especially in infants. Certain occupations, including painting, glazing, varnishing, lithography, and printing, are high risk for inhalational exposure to methanol. Inhalational abuse of methanol is a recent trend that can result in toxic serum levels.2 Methanol itself has little toxicity, producing less central nervous system (CNS) depression and inebriation than ethanol. Metabolites of the parent alcohol are extremely toxic, however. Although small amounts of methanol are eliminated by renal and pulmonary routes, 90% is metabolized in the liver. Methanol is oxidized by alcohol dehydrogenase (ADH) to formaldehyde, which is rapidly converted by aldehyde dehydrogenase to formic acid (Fig. 155-1). Formic acid is the primary toxicant and accounts for much of the anion gap metabolic acidosis and ocular toxicity peculiar to methanol ingestion.3 Through a folate-dependent pathway, formic acid is degraded to carbon dioxide and water. Optic neuropathy and putaminal necrosis are the two main complications of severe methanol poisoning. Long-term morbidity takes the form of visual impairment, including blindness, and parkinsonian motor dysfunction, characterized by hypokinesis and rigidity. Formic acid has a high affinity for iron and inhibits mitochondrial cytochrome oxidase, halting cellular respiration.4 Methanol metabolism in the cytosol and mitochondria may account for a second mechanism of adenosine triphosphate depletion.5 Lactate accumulation resulting from hypotension or seizures further compounds the metabolic acidosis predominantly caused by formate. Other mechanisms of toxicity involve increased lipid peroxidation, free radical formation, and impaired protective antioxidant reactions.4,6 Severe dysfunction of subcellular metabolism from methanol also has been linked to significant disturbance in proteolytic-antiproteolytic balance.7 The primary sites of ocular injury are the retrolaminar optic nerve and retina. Selective myelin damage to the retrolaminar optic nerve has been seen at autopsy after methanol toxicity. Müller cells, the principal glial cells of retinal neurons and photoreceptors, have been proposed as the initial target in methanol-induced visual toxicity. It seems that they alone harbor the enzymes necessary to metabolize methanol to formate. Histopathologic correlates suggest that retinal cells develop intra-axonal swelling, calcium influx, mitochondrial destruction, and microtubular disruption. Ultimately, this interferes with transport of essential proteins from the retinal neuron cell body to the nerve fiber axoplasm. Oligodendrocyte involvement causes myelin degeneration and leads to visual decrements. Acidosis may accelerate this process by enhancing nonionic diffusion of formic acid into neurons and further increasing lactate production.4 This self-perpetuating cycle of acidosis, termed circulus hypoxicus,8 underscores the need for aggressive correction of pH to accomplish ion trapping of formate outside the CNS. Methanol adversely affects other areas of the CNS, specifically the basal ganglia. Bilateral, symmetrical putaminal hypodensities, hemorrhages, or cystic lesions are characteristic, occurring in 13.5% of patients. Necrosis is described in the subcortical white matter, spinal cord anterior horn cells, and cerebellum.9 Acute signs and symptoms may be lacking or may take several days to develop despite the presence of these radiographic findings. The cellular mechanisms of injury may be similar to the mechanisms of the ophthalmologic injury, but the reason for localization of neurologic damage to the basal ganglia is unknown. Quantitative neuropathologic studies conflict as to whether concentrations of formic acid within the putamen are higher than levels in the blood or other tissues. Massive edema adjacent to the putamen shown by magnetic resonance imaging (MRI) suggests a possible localized disruption of the blood-brain barrier. Other proposed mechanisms for the vulnerability of this region include the unique pattern of arterial blood supply and venous drainage and greater metabolic activity. With individual cases of methanol poisoning, the history may be unobtainable or unreliable. The diagnosis should be considered in patients with altered mental status, visual complaints, or metabolic acidosis and in patients with occupations that put them at high risk for exposure. Because methanol is a poor substrate for ADH, a latency period exists between the time of ingestion and metabolism to the formate that causes visual or metabolic disturbance. The typical 12- to 24-hour latency may be shorter when large amounts are consumed or longer when ethanol is coingested (range, 40 minutes to 72 hours).1 Formic acid accumulation may be ongoing, with risk for significant toxicity, in patients who present early despite being initially asymptomatic. When symptoms are manifested, they are primarily neurologic, gastrointestinal, or ocular. Although methanol is less inebriating than ethanol, early symptoms of methanol poisoning include depressed mental status, confusion, and ataxia. Nonspecific complaints of weakness, dizziness, anorexia, headache, and nausea develop; in severe cases, coma and seizures may be seen. Although vomiting and abdominal pain commonly result from mucosal irritation, the absence of gastrointestinal complaints does not rule out a serious ingestion.1 Abdominal tenderness, however, may be so severe that it suggests an acute surgical abdomen.1 This may result from pancreatitis, and elevation of serum amylase is relatively common, but increased salivary amylase isoenzyme can occur without pancreatic inflammation. Visual disturbances are seen in 50% of patients, and their development may precede or parallel that of other symptoms.8 Patients may complain of cloudy, blurred, indistinct, or misty vision or may note yellow spots or, rarely, photophobia. The most common acute field defect is a dense central scotoma.10 Some patients compare their visual symptoms with “stepping out into a snowstorm,” a complaint unique to methanol ingestion. Patients can have a complete lack of light perception and total loss of vision. Visual acuity should be determined. On examination, optic disc hyperemia is seen at 18 to 48 hours after ingestion. Peripapillary retinal edema follows, is most striking in the nerve fiber layer along the vascular arcades, and only rarely involves the macula.5,10 Sluggishly reactive or fixed and dilated pupils indicate a poor prognosis. Pallor and cupping, indicative of optic atrophy, are late findings suggesting a poor prognosis for visual recovery. On occasion, the fundus may appear normal, even in patients with visual symptoms. Compensatory tachypnea heralds the onset of metabolic acidosis, which often may be severe, with reported serum bicarbonate concentrations of less than 5 mEq/L and an arterial pH less than 7.0. Early tachycardia has been noted, but in general, cardiovascular abnormalities are rare.1 Hypotension and bradycardia, when present, are preterminal findings.11 Historically, death is associated with a peculiar, abrupt cessation of respiration rather than with cardiovascular collapse.1 Multiple organ failure is rare.11 Prognosis after methanol ingestion seems to correlate with the degree of acidosis, time to presentation, and initiation of treatment within 8 hours of exposure.1,12 Poor prognosis is associated with coma, seizures, or arterial pH less than 7.0.11 A large outbreak was associated with a fatality rate of 44%.13 Patients surviving the acute phase of toxicity may be left with permanent blindness or neurologic deficits, such as parkinsonism, toxic encephalopathy, polyneuropathy, cognitive dysfunction, transverse myelitis, primitive reflexes, or seizures.14 The “normal” osmol gap is often cited to be less than 10 mOsm/kg when the preceding equation is used. This is an arbitrary number, and there is considerable variability in baseline osmolal gaps in patients, particularly children. An osmol gap significantly greater than 10 mOsm/kg may be a useful aid in the diagnosis of toxic alcohol ingestion. In addition to methanol, ethylene glycol, and isopropanol, other low-molecular-weight solutes, such as ethanol, acetone, propylene glycol, mannitol, glycerol, and ethyl ether, may cause elevated osmol gaps. Caution should be taken, however, in ruling out toxic alcohol ingestion with a normal osmol gap for several reasons. First, calculated serum osmolality results may vary among laboratories and must be done by the freezing point depression method. Also, delayed presentation after toxic alcohol ingestion may be associated with prior metabolism of most of the parent alcohol. Because only the parent compound is osmotically active and because the charged metabolites are electrically balanced by sodium, there may be little or no osmol gap elevation in this setting. Finally, a toxic level of either methanol or ethylene glycol may be present with a gap of only 10 mOsm/kg. If there is clinical suspicion of toxic alcohol ingestion, direct measurement of the serum toxic alcohol level is necessary, and if it is not readily available, empirical treatment is warranted.15 Rhabdomyolysis, pancreatitis, and metabolic derangements, such as hypomagnesemia, hypokalemia, and hypophosphatemia, are also described with methanol poisoning. Computed tomography may be indicated in an intoxicated patient with altered mental status. The characteristic finding of bilateral putaminal lesions suggests methanol poisoning, but this finding also may be seen with Leigh’s syndrome, Wilson’s disease, hypoxic-ischemic insult, encephalitis, and certain metabolic disorders. Ischemic necrosis, cerebral edema, or brain hemorrhages also may be noted. Follow-up scans may have prognostic value because parkinsonian features are unlikely to develop in patients whose putaminal lesions resolve within a short time frame.16 MRI may also detect putaminal aberrations or optic neuropathy from methanol intoxication. Ethylene glycol is a viscous, colorless, odorless, slightly sweet-tasting liquid. Because it lowers the freezing point of water, its primary utility is as a commercial antifreeze or coolant. Other sources include airplane deicing solutions, hydraulic brake fluids, and industrial solvents and precursors; it also is a component of certain paints, lacquers, and cosmetics. Most ethylene glycol poisonings occur with the ingestion of antifreeze. Unusual poisoning scenarios are described, including an epidemic after the contamination of water supplies and the intentional poisoning of an infant, manifested as an inherited metabolic disorder. In 2009, the American Association of Poison Control Centers reported 5977 exposures to ethylene glycol. Of those exposures, 72% were unintentional, and 11% resulted in moderate or severe effects with 19 fatalities. If it is treated early and aggressively, ethylene glycol poisoning is unlikely to result in death. Conversely, failure to treat ethylene glycol ingestion may result in multiorgan failure and death within 24 to 36 hours.17 Absorption of ethylene glycol is rapid after ingestion. It distributes evenly to tissues with a volume similar to that of body water. Peak blood levels are reached within 1 to 4 hours after ingestion. In contrast to methanol and isopropanol, ethylene glycol is nonvolatile at room temperature, so absorption by inhalation is unlikely. Reported half-lives range from 3 to 8.6 hours.18 When metabolism is blocked by fomepizole or ethanol, the half-life increases to 11 to 15 hours or 17 hours, respectively.19 The toxic and lethal doses of 100% ethylene glycol have been reported as 0.2 mL/kg and 1.4 mL/kg. At the other extreme, with modern treatment, patients who have ingested 3000 mL have survived. Twenty-seven percent of ethylene glycol is excreted unchanged by the kidneys. The remainder is oxidized through hepatic ADH and other oxidative enzymes to various toxic organic aldehydes and acids (Fig. 155-2). Unmetabolized ethylene glycol has limited toxicity, yet all metabolites are toxic. In humans, 2.3% of a dose of ethylene glycol ultimately is converted to oxalic acid, most of which is excreted in the urine. A fraction of oxalic acid combines with calcium to form calcium oxalate crystals, which precipitate in renal tubules, brain, and other tissues. Studies have shown definitively that the accumulation of calcium oxalate monohydrate crystals in kidney tissue produces renal tubular necrosis that leads to kidney failure.20 Other authors suggest that glycolate levels correlate better with disruption of CNS metabolism, development of renal failure, and mortality.21 Because the intermediate metabolite, glyoxylic acid, theoretically can be shunted toward pyridoxine-dependent or thiamine-dependent pathways to generate the nontoxic products glycine and α-hydroxy-β-ketoadipate (see Fig. 155-2), both pyridoxine and thiamine are routinely used in therapy. On histologic examination, proximal renal tubular dilation with hydropic change and vacuolar degeneration, intratubular crystal deposition, and edema of the interstitium are seen. Relative sparing of the glomeruli is typical, although glomerular interloop space crystal deposition may occur. Glomerular function is preserved, but tubular dysfunction manifesting as protein leak is noted.22 Neuropathologic changes induced by ethylene glycol include diffuse calcium oxalate deposition with petechial hemorrhages in the retina, brain, vessel walls, and perivascular spaces, with evidence of cerebral edema and chemical meningoencephalitis. Similar changes also have been noted in the liver, spleen, pancreas, pleura, lungs, pericardium, and blood vessel walls throughout the body. Myonecrosis occurs in skeletal and myocardial muscle, and rhabdomyolysis and myocardial dilation occur. Fatty liver with focal necrosis has been noted.
Toxic Alcohols
Methanol
Principles of Disease
Pathophysiology
Clinical Features
Diagnostic Strategies

Ethylene Glycol
Principles of Disease
Pathophysiology

Full access? Get Clinical Tree


Toxic Alcohols
Only gold members can continue reading. Log In or Register to continue




