1. The development of the urogenital system can conceptually be divided into its urinary portion and genital portion.
2. Specific anesthetic considerations for patients with Wilms tumor include a thorough imaging evaluation to include tumor extension to the renal vein or inferior vena cava (IVC), local (mass) effects on contiguous structures like the gastric outlet, systemic effects (anemia, thrombocytopenia, hypertension), and the effects of preoperative chemotherapy.
3. Bladder exstrophy (epispadias) and cloacal anomaly patients may require many reconstructive procedures and may have associated ectodermal hindgut anomalies such as myelomeningocele in complete or incomplete forms, including tethered cord. They may also be genomically and environmentally susceptible to natural rubber latex allergy.
4. Vaginoplasty or clitoroplasty patients may have congenital adrenogenital hyperplasia with cortisol and aldosterone insufficiency and androgen excess and are typically on replacement gluco- and mineralocorticoids.
5. Nephrogenic Diabetes Insipidus (NDI) is a disorder characterized by the inability to concentrate urine due to a lack of response of renal tubules to antidiuretic hormone.
DEVELOPMENT OF THE UROGENITAL SYSTEM
THE URINARY PORTION OF THE UROGENITAL SYSTEM
Three structures—the pronephros, mesonephros, and metanephros—appear sequentially in development and are responsible for differentiating into the kidneys and ureters—the urinary portion of the urogenital system (Fig. 25.1). The pronephros develops early in the 4th week from mesodermal cells in the cervical region and regresses by the end of the 4th week. The mesonephros along with the mesonephric ducts form from mesoderm in the thoracic and lumbar regions at about the same time as the evanescent pronephros. Mesonephric excretory tubules are formed and lengthen, forming a loop crowned by capillary growth that will eventually become the glomerulus, surrounded by Bowman capsule medially, and will enter the lateral aspect of the mesonephric (Wolffian) duct. The metanephros (definitive kidney) begins its development in the 5th week. The kidney’s collecting ducts develop from the ureteric bud, an outgrowth of tissue at the caudal end of the mesonephros next to the cloaca, which invests the surrounding metanephric tissue. Local tissue growth develops into the renal pelvis and subsequently the major renal calyces. The formation of minor calyces is a joint effort of outgrowing major calyces and ingrowing collecting tubules, eventually forming renal pyramids. Capped by metanephric tissue, the collecting tubule forms the renal vesicles that subsequently develop into glomeruli. The tubule plus glomerulus form a nephron, connected at its distal end to the collecting tubule. This tubule lengthens and loops, forming the proximal convoluted tubule, the loop of Henle, and the distal convoluted tubule.
Abnormalities of this sequence of development may lead to tumors (Wilms tumor, or nephroblastoma, resulting from a mutation of the WT1 gene on 11p13), or syndromes associated with tumors (WAGR syndrome—aniridia, hemihypertrophy, and Wilms tumor; Denys-Drash syndrome—renal failure, pseudohermaphroditism, and Wilms tumor). Dysplastic/polycystic kidneys can result when cells do not continue to differentiate into nephron units; normally developed ducts may be surrounded by dysplastic and undifferentiated cells. In severe forms, renal agenesis may be the result. Potter Syndrome is the result of bilateral renal agenesis.
Abnormalities of the collecting system such as a double collecting system/ureteral duplication (Fig. 25.2) result from segmentation of the ureteric bud; metanephric tissue may develop into two renal pelves and ureters. Ureters may empty into adjacent organs as well, such as the vagina or urethra. The second ureteric bud typically moves caudally with the mesonephric duct, and therefore will communicate with adjacent organs at a more caudal site.
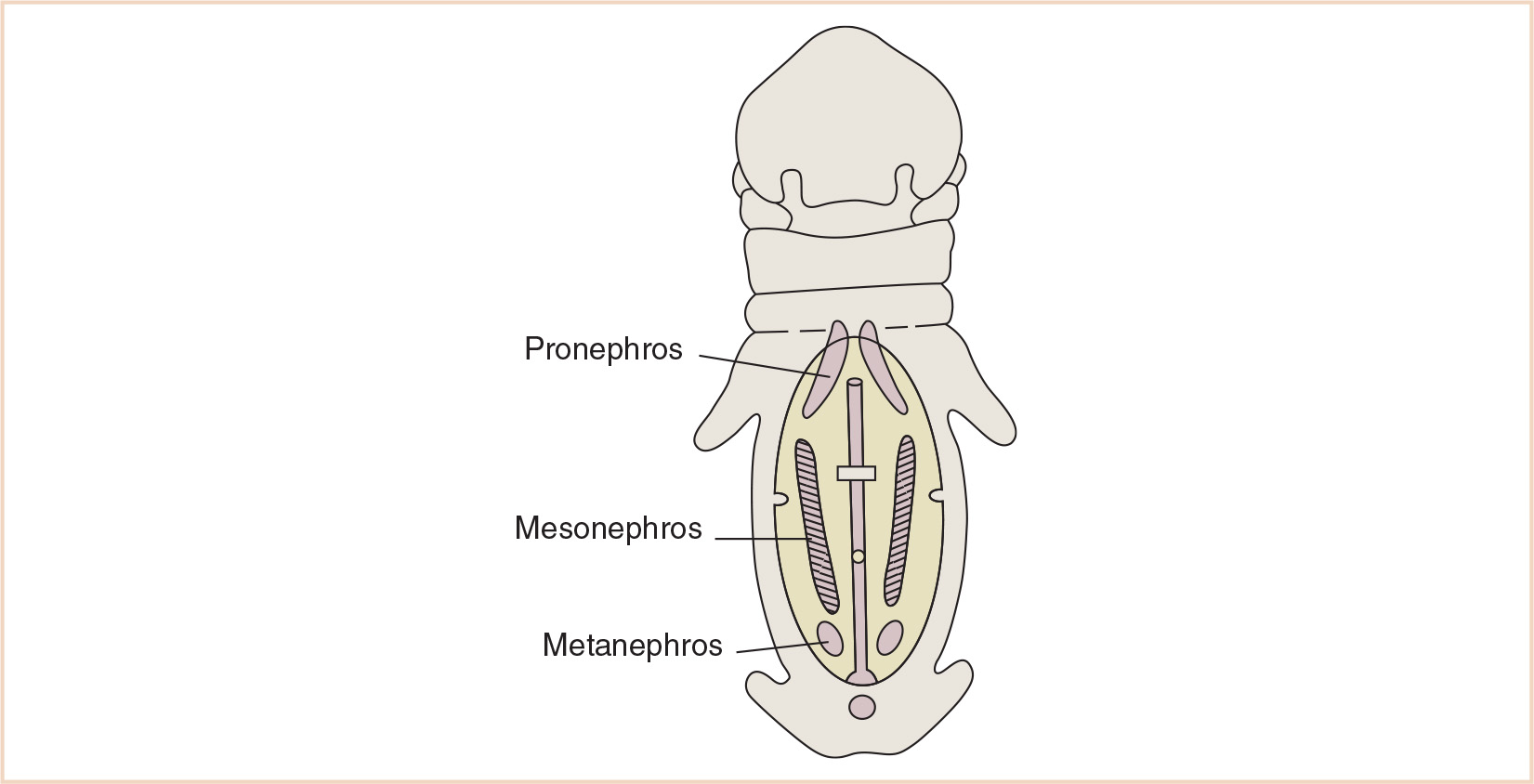
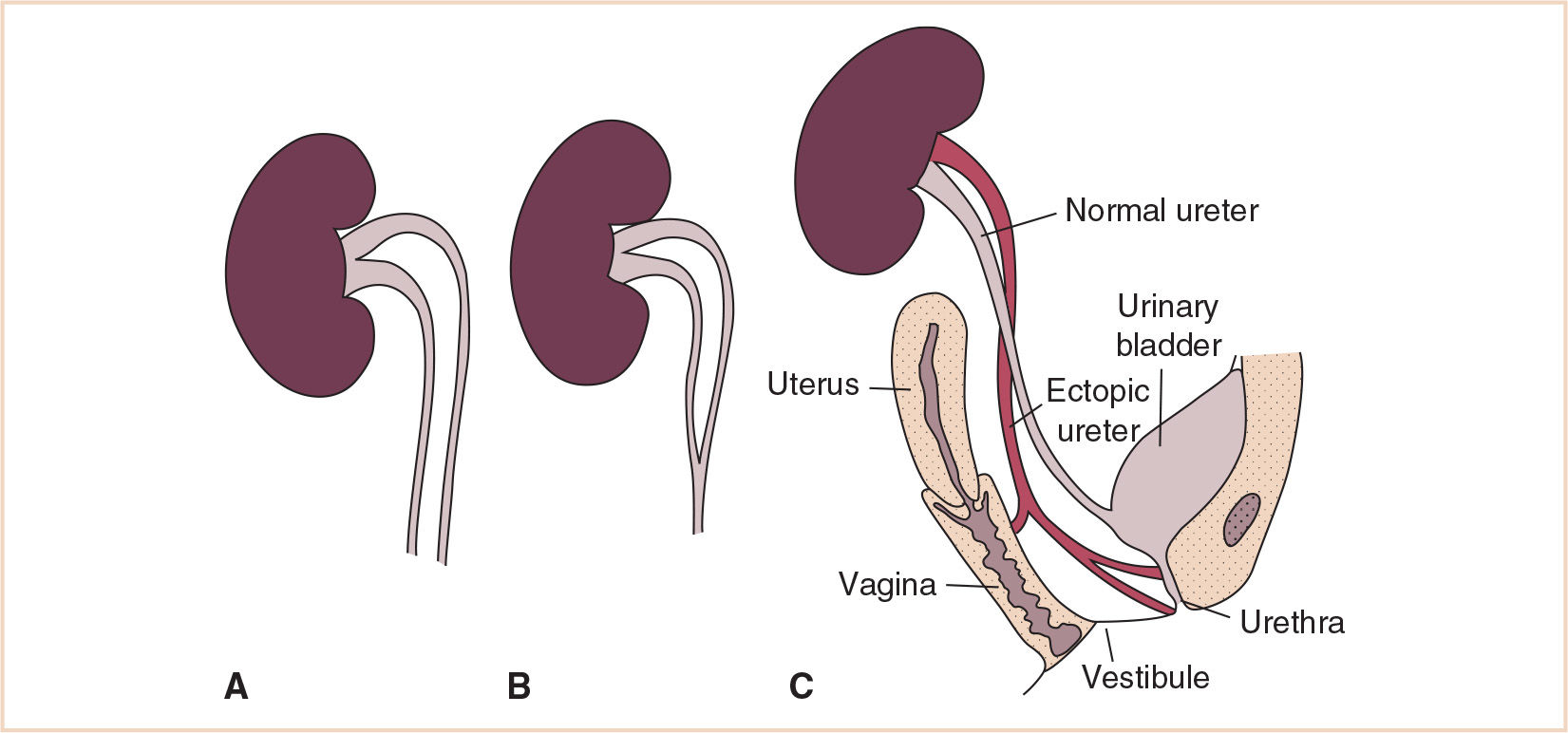
FIGURE 25.2 Duplicated collecting system illustrating a variety of emptying locations for the distal ureter. (A) Double pelvis (B) Bifid ureter (C) Ectopic ureteric orifice.
Because the kidney begins as a pelvic structure, its migration is subject to abnormalities of growth and development as well. One or both kidneys can remain in the pelvis or they can fuse to form a horseshoe kidney.
The cloaca undergoes division by caudal growth of the urorectal septum to become the anterior urogenital sinus and the posterior anal canal (Fig. 25.3). The caudal-most portion of the mesodermally-derived urorectal septum becomes the perineal body. The bladder is formed from the portion of the allantois closest to the cranial portion of the urogenital sinus; the balance of the allantois undergoes obliteration but remains as a fibrous band connecting the cranial portion of the bladder to the umbilicus; this ligamentous connection is called the urachus in the fetus and the median umbilical ligament postnatally (Fig. 25.4). The middle portion of the urogenital sinus (in the male) becomes the prostatic urethra and the caudal-most portion the phallic portion of the urogenital sinus, not having undergone gender differentiation as yet.
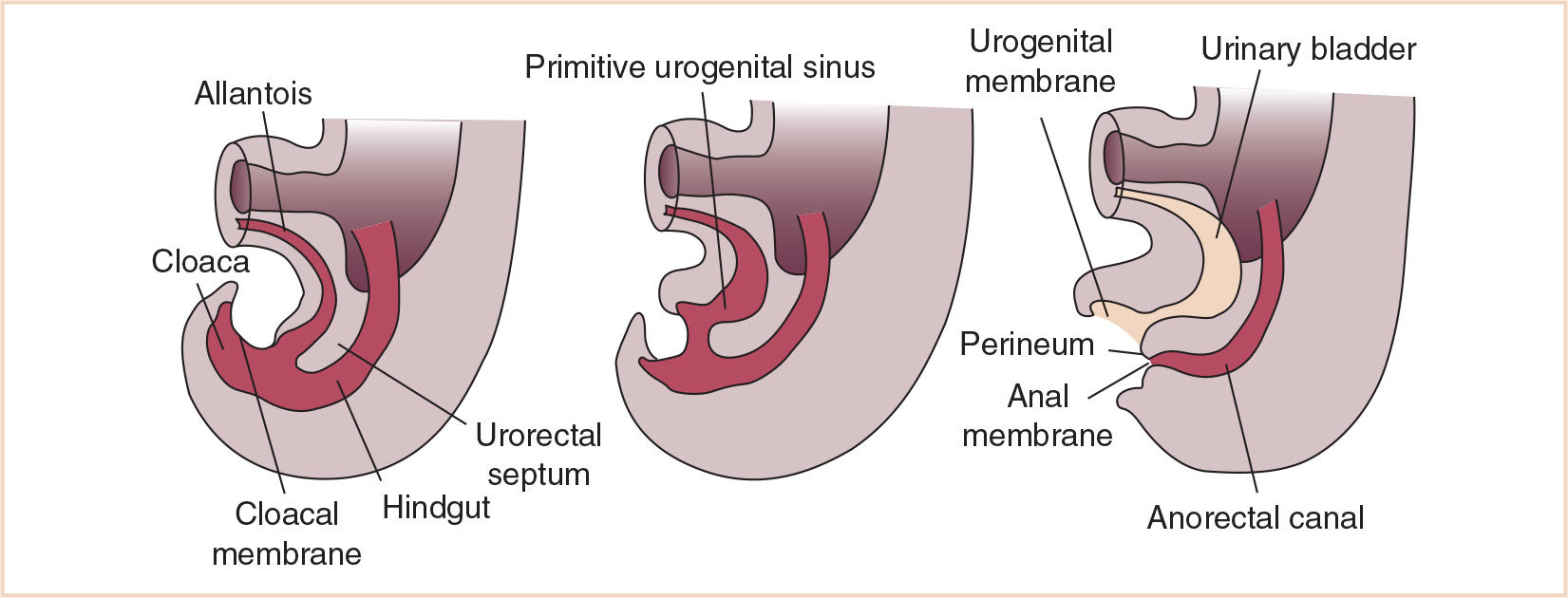
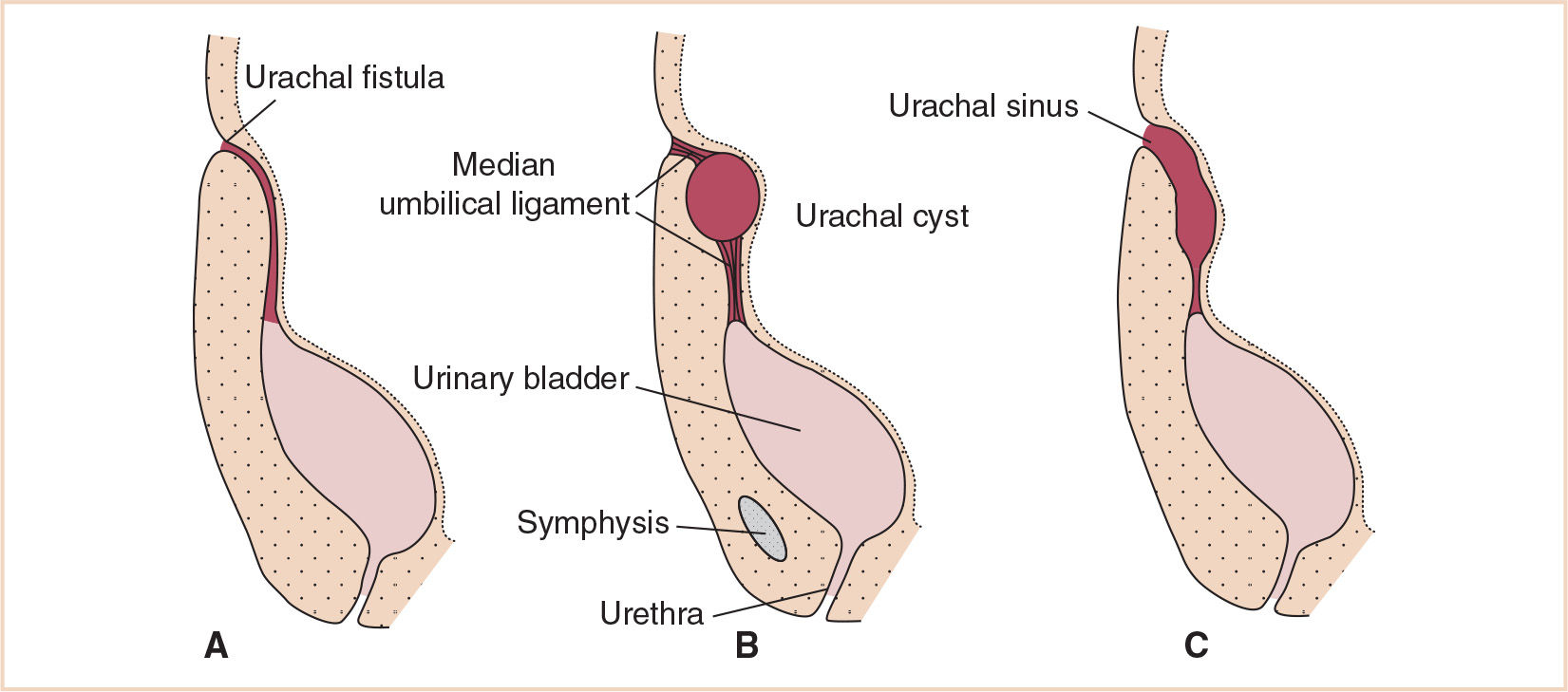
FIGURE 25.4 Abnormal remnants of a patent urachus. (A) Urachal fistula (B) Urachal cyst (C) Urachal sinus.
The caudal portions of the mesonephric ducts are incorporated into the lateral wall of the developing bladder. Kidney ascent cranially results in the ureteral orifices moving cranially as well, while the orifices of the mesonephric ducts move closer together to enter the prostatic urethra, becoming the ejaculatory ducts.
Bladder defects occur with variable severity. Defects in obliteration of the urachus may result in a urachal fistula, cyst, or sinus anywhere between the abdominal wall and the bladder. In more extreme forms, bladder exstrophy is a body wall defect exposing the bladder mucosa, with the urinary tract defect extending along the dorsum of the penis. Cloacal exstrophy is a more extreme form of body wall defect where the bladder mucosa is exposed in association with other hindgut and ectodermal abnormalities such as imperforate anus, meningomyelocele, and possibly an omphalocele.
THE GENITAL PORTION OF THE UROGENITAL SYSTEM
The gonads start to take on a male and female appearance during the 7th week. While initially emerging as paired retroperitoneal gonadal ridges, germ cells do not begin to invest this mesenchyme until the 6th week, when they arrive after their migration from the caudal portion of the yolk sac in the region of the allantois. Failure of this migration results in failure of gonadal development. Even with the arrival of these primordial germ cells, and cell signaling resulting in penetration of underlying mesenchyme by overlying epithelial cells, the ensuing primitive sex cords are neither male nor female but rather, at this point, are known as indifferent gonads.
If genetically male, the primordial germ cells will signal the primitive sex cords and form the medullary cords of the testis that will eventually (in the 4th month) develop into the primitive germ cells. Meanwhile, the interstitial cells of Leydig, having developed from the gonadal ridge mesenchyme, begin to produce testosterone during the 8th week and promote the sexual differentiation of the genital ducts and external genitalia.
If genetically female, the primitive sex cords break up into clusters that contain the germ cells that will reside in the medullary portion of the ovary, eventually degenerating, to be replaced by the ovarian medulla. The surface epithelium of the female gonad will continue to grow and will eventually form the cortical cords, which, like the primitive sex cords, will also break up and be invested with germ cells, subsequently developing into oogonia and the epithelially-derived follicular cells.
Genital ducts during the indifferent stage in both genders consist of two pairs: the mesonephric (Wolffian) ducts and the paramesonephric (Müllerian) ducts. The paramesonephric duct runs lateral to the mesonephric duct then crosses it, fusing with the paramesonephric duct from the opposite side to form the uterine canal; the septum between the two degenerates during the normal process. This development, moreover, is under the influence of estrogens in the developing female embryo. The paramesonephric ducts are stimulated to form the tubes of the uterus, the uterus itself, the cervix, and the proximal vagina. In addition, the labia majora, minora, clitoris, and distal vagina are under the developmental influence of estrogens.
The testes descend from the posterior abdominal wall in the region of the mesonephros; the caudal genital ligament, a remnant of the urogenital mesentery which attached the testis and mesonephros to the posterior abdominal wall, becomes confluent with the gubernaculum, the trajectory along which the testis descends to the scrotum. The testis normally progresses to the inguinal canal by the 12th week, through the inguinal canal by the 28th week and into the scrotum by the 33rd week. The blood supply from its mesonephros origin (lumbar vessels) is retained as the testis migrates to the scrotum. A pouch of peritoneum, the processus vaginalis, develops along the path of the gubernaculum into the scrotum, along with the muscle and fascial layers from the anterior abdominal wall. These are the contents of the inguinal canal.
Abnormalities from impairment of these steps can result in a persistent communication via the processus vaginalis, a congenital inguinal hernia. Residual cysts in this path without an overt hernia will present as hydroceles (Fig. 25.5). For the 1% of male infants with arrest of testicular descent, cryptorchidism is the result.
The body and cervix of the uterus form from the fused caudal ends of the paramesonephric ducts while the vagina is formed from the wall of the urogenital sinus (Fig. 25.6). The vaginal plate is an outgrowth of tissue from the urogenital sinus. The vaginal plate elongates and tissue breakdown occurs within it, with the vaginal lumen remaining separated from the lumen of the urogenital sinus by the hymen.
Abnormalities in formation of the uterus and vagina from these paired structures can be the result of fusion failures, stenoses, or atresias, and result in a double uterus–double vagina (uterus didelphys), bicornuate uterus (with a normal vagina), or a normal uterus with vaginal atresia.
Male genital abnormalities also typically arise from failures of fusion of paired structures, such as hypospadias (from failure of the urethral folds to fuse in the midline with abnormal openings along the inferior aspect of the penis). The occurrence is approximately 33:10,000 in the United States; fathers of children with hypospadias are affected in 8% and brothers are affected in 14%. Epispadias is much more rare, with the urethral meatus emerging on the dorsum of the penis, often associated with bladder exstrophy.
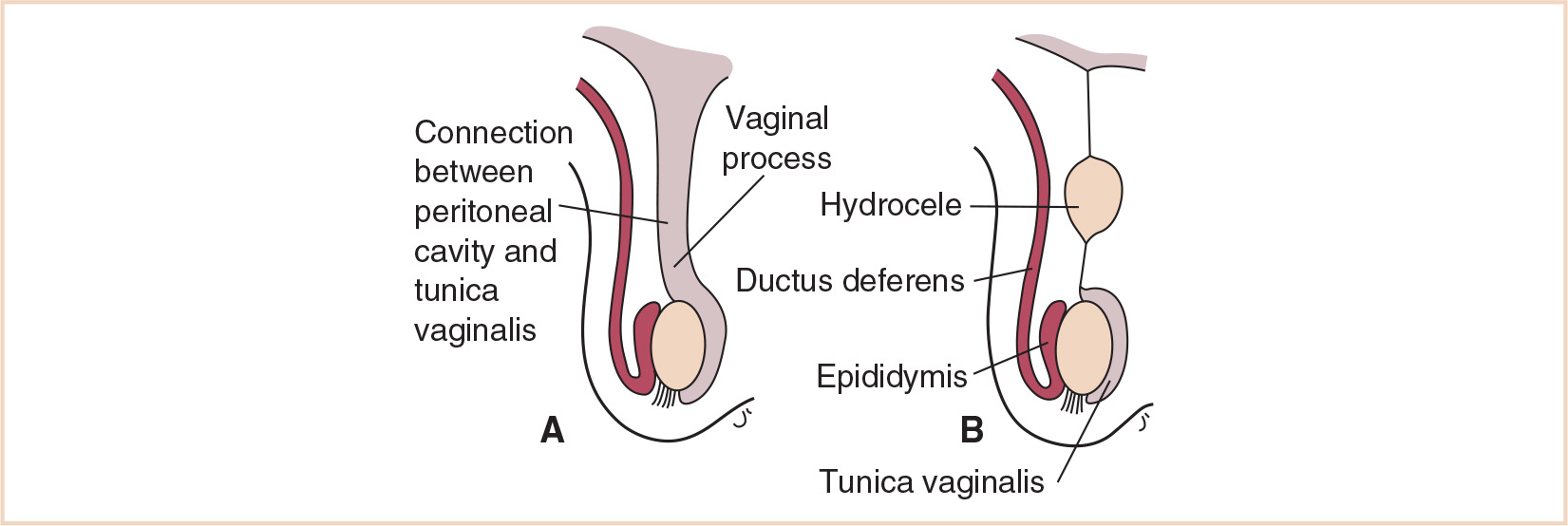
FIGURE 25.5 A patent processus vaginalis results in a hernia (A); a cyst within an obliterated processus vaginalis is a hydrocele (B).
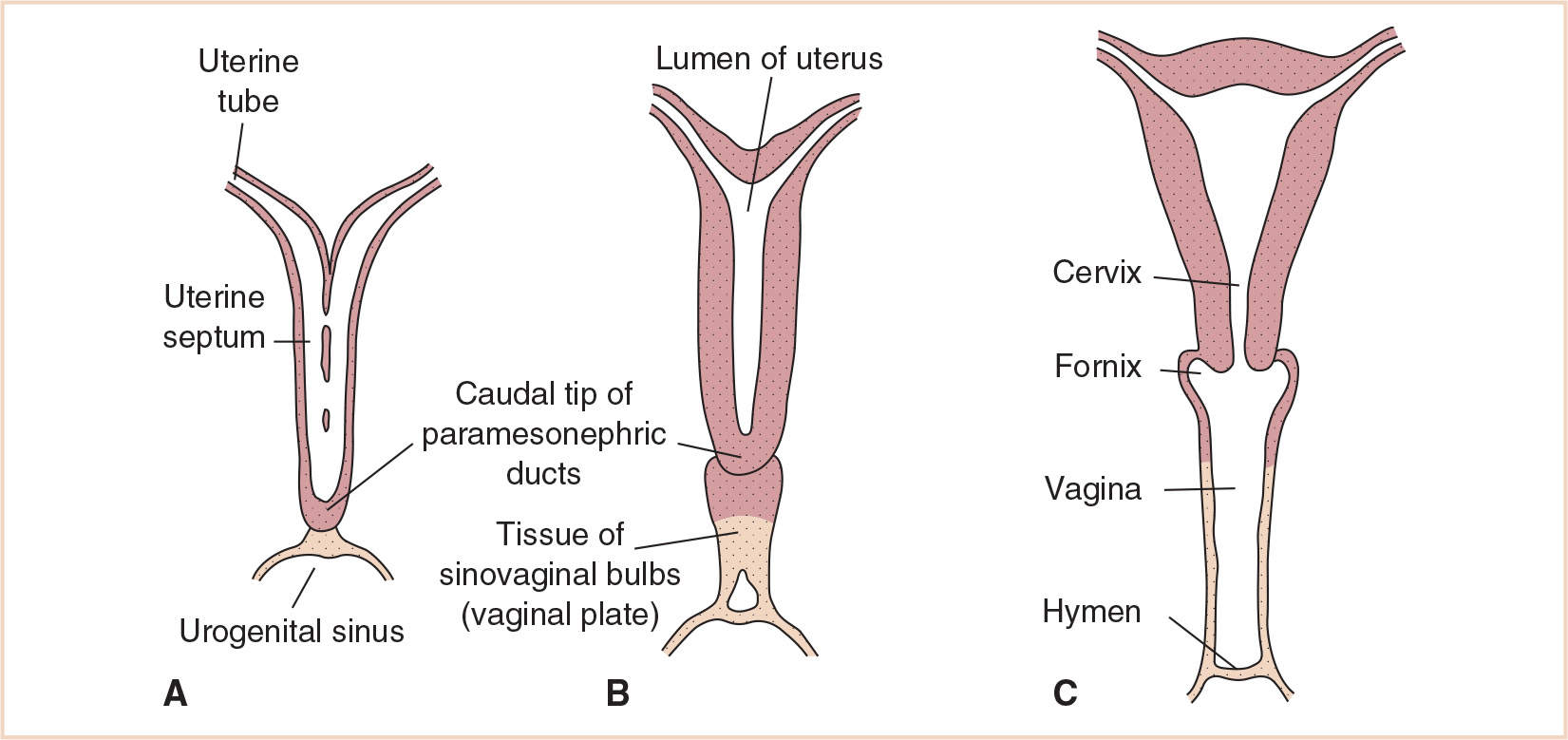
FIGURE 25.6 Formation of the body of the uterus from the fused caudal portions of the paramesonephric ducts with (A) and later without (B) a median septum. (C) The vagina is formed from the wall of the urogenital sinus and the hymen from the distal portion of the vaginal plate and the wall of the urogenital sinus.
DISORDER: Kidney abnormalities (congenital anomalies; dysfunctional kidneys from reflux nephropathy; symptomatic polycystic kidney disease); duplicated system, ectopic ureter, or ureterocele; upper pole obstruction of a duplex system; hydronephrosis due to obstruction of the ureteropelvic junction; abnormalities of the distal ureter with impaired urine flow (failed ureteral reimplant, conversion to a single emptying system prior to a diversion procedure, treatment for ureteral trauma)
EMBRYOLOGY/ANATOMY
The kidney develops from the metanephros. The collecting ducts of the kidney develop from the ureteric bud at the caudal end of the mesonephric duct, which invests the metanephric mesoderm in the pelvis. The resulting metanephric blastema ultimately differentiates into the nephron and “ascends” (or appears to ascend) cranially, eventually settling in the lumbar area as the body grows in the lumbar and sacral regions. Abnormalities of blastema differentiation may result in dysplastic or polycystic kidneys, or congenital nephroblastoma (Wilms tumor). Complete and partial duplications of the ureter may occur anywhere along the path of the ureteric bud and the mesonephric duct. Abnormalities of the ureteric bud itself can result in dysplasia/ectasia of the renal pelvis or implantation abnormalities of the distal ureter.
PHYSIOLOGIC CONSIDERATIONS
Pediatric kidney and urinary tract disease requiring surgical intervention may be accompanied by any of the following:
1. Hypertension
2. Pyelonephritis
3. Renal insufficiency or renal failure (acute or chronic)
4. Pain
5. Secondary effects of cancer chemotherapy
6. Failure to thrive
7. Psychological issues due to treatment for chronic urinary tract infections and/or repeated diagnostic procedures
8. Anemia of chronic disease
SURGICAL REPAIR: Radical nephrectomy/simple nephrectomy/partial nephrectomy/nephrou-reterectomy/pyeloplasty/transureteroureterostomy (TUU)
1. Radical nephrectomy: Wilms tumor (nephroblastoma, embryoma of the kidney) is congenital although it does not typically present clinically until about 3 years of age. With a multidisciplinary approach incorporating chemotherapy with vincristine, dactinomycin (actinomycin D), and doxorubicin, overall survival has risen to 90% (1). Most cases are not part of a genetic malformation syndrome and have no familial history; however, familial Wilms tumor arises with high frequency in certain families. Genetic syndromes that predispose to and may include Wilms tumor include: Beckwith-Wiedemann syndrome (macroglossia, gigantism, and umbilical hernia), hemihypertrophy, congenital aniridia, WAGR syndrome (Wilms tumor, aniridia, genitourinary malformations, and mental retardation), Denys-Drash syndrome (Wilms tumor, pseudohermaphroditism, and glomerulopathy) and Trisomy 18. Indications for primary surgical excision of a Wilms tumor include tumors confined to the kidney, extending beyond the kidney but not crossing the midline, and those with or without vascular extension (Table 25.1). Intravascular extension (inferior vena cava, right atrium) occurs in 4% to 6% and is frequently asymptomatic (2). Postchemotherapy excision of the tumor is indicated in patients with bilateral tumors, tumors that extended beyond the midline but have shrunken, and tumors with vascular extension. Surgery alone is not recommended for Wilms tumor. The tumor extends into the renal vein in 40% of cases, although it rarely extends into the ureter and down to the bladder. It is bilateral in 6% of cases. Intracaval tumor extension occurs in 5% and has a 40% rate of surgical complications. Presurgical chemotherapy after staging and biopsy reduces tumor and thrombus size, which account for 25% of surgical complications.
2. Simple nephrectomy: In children, nephrectomy is typically performed for congenital anomalies or chronic recurrent infection with significantly impaired renal function and secondary consequences (recurrent pyelonephritis, hypertension). Polycystic kidney disease may also be associated with pain.
3. Partial nephrectomy: Usually performed for the nonfunctioning upper pole of a duplicated collecting system. Resection of the upper collecting system may include reanastomosis with the lower pole collecting system via pyeloureterostomy.
4. Nephroureterectomy: Typically performed for ureterocele or ectopic ureter in a duplicated collecting system. The ureter is resected as distal as the incision will allow, usually to the level of the iliac vessels.
5. Pyeloplasty: Performed for hydronephrosis, which is often due to ureteropelvic junction (UPJ) obstruction for congenital reasons. The UPJ is resected with a primary reanastomosis of the ureter and renal pelvis.
6. Transureteroureterostomy: Used to decompress and divert one obstructed ureter through resection and reanastomosis to the opposite ureter. May be used following a failed reimplantation or in cases of neurogenic bladder with the creation of a diversion. An abdominal incision (midline or Pfannenstiel) is the open surgical approach; TUU is also performed laparoscopically by conventional means or by robotic assistance.
TABLE 25.1 Staging of Wilms tumor
1 | The tumor is limited to the kidney and is excised completely. |
2 | The tumor extends beyond the kidney but is excised completely. Capsular penetration, renal vein involvement, and renal sinus involvement also may be found. A biopsy of the tumor is performed, and local spillage occurs. |
3 | Residual intra-abdominal tumor (nonhematogenous) exists after the completion of surgery. Lymph node findings are positive, or peritoneal implants are found. The resected specimen has histologically positive margins, or the tumor has been spilled into the abdominal cavity. |
4 | Hematogenous or lymph node metastasis has occurred outside the abdomen or pelvis. |
5 | Synchronous bilateral involvement has occurred. Each side is assigned a stage from I to III, and histology is based on biopsy findings. |
CLINICAL PEARL With a multidisciplinary approach incorporating chemotherapy with vincristine, dactinomycin (actinomycin D), and doxorubicin, overall survival has risen to 90% (1). Most cases are not part of a genetic malformation syndrome and have no familial history; however, familial Wilms tumor arises with high frequency in certain families.






Full access? Get Clinical Tree


