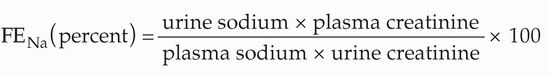The kidneys help preserve intravascular volume, excrete products of metabolism, and regulate acid-base status. The physiology of sodium and water reabsorption that is linked to renal blood flow engages surgical critical care clinicians on a daily basis. Urine output is commonly used as a substitute for measurement of renal perfusion and as a surrogate for cardiac output monitoring. Acid-base disturbances are also a daily feature of surgical critical illness. Acute kidney injury (AKI) regularly accompanies shock from hypoperfusion and/or systemic inflammation. Avoidance of and/or limiting the duration of AKI is linked to the avoidance and/or limitation of shock and, therefore, is an important strategy to improve outcome in surgical critical illness.
Selected aspects of renal physiology provide the foundation for the concepts of evaluation and management of renal function that are pertinent to surgical critical illness.
Glomerular Filtration
The first process in the formation of urine is glomerular capillary ultrafiltration of blood. This results in a fluid that is low in protein and contains all solutes not bound to non-filterable proteins.
GFR (the amount of blood filtered/minute) is measured by determining the clearance from the plasma of a substance that is filtered but neither secreted nor absorbed by the renal tubules. Dividing the amount excreted in the urine over a given amount of time by the plasma concentration of that substance in normal adult humans results in a GFR of 100-125 cm
3/min (144-160 L/day) (
2).
GFR is influenced by a variety of mechanism that attempt to maintain filtration despite the potential of marked alterations in renal blood flow (
Table 7.1) (
3,
4).
Tubular Reabsorption
The 100-125 cm
3/min of glomerular filtrate enters the proximal renal tubule, which, along with the rest of the tubular system, reabsorbs about 98% of the filtered water and solutes. The proximal convoluted tubule (PCT) reabsorbs approximately 60% of overall glomerular filtrate, with greater percentages of phosphate, water and bicarbonate (80%), and glucose and amino acids (100%). The thick ascending limb of Henle absorbs approximately 30% of the filtrate with the distal convoluted tubule responsible for about 7% and the collecting ducts absorb the remaining few percent (
3,
5).
Sodium Reabsorption
The reabsorption of sodium is the major active transport mechanism of the entire nephron. The three major determinants of sodium reabsorption are listed in
Table 7.2. GFR influences the active process of sodium reabsorption in the proximal tubule, such that an increase in filtration is associated with an increase in total absorption (glomerular-tubular balance). This process appears to be independent of direct neurohumoral control (
2,
5).
Proximal tubule absorption is augmented by adrenergic innervation and low-dose angiotensin as well as insulin and glucocorticoids. Dopamine inhibits proximal sodium absorption and this effect can by augmented by natriuretic hormones—atrial natriuretic peptide (ANP) and urodilatin (Urod) (
5,
6).
In the thick ascending loop of Henle, glucocorticoids, vasopressin, beta adrenergic stimulation, and insulin augment absorption, whereas dopamine, ANP, and Urod are inhibitory (
5).
Aldosterone augments sodium absorption and is released as the end product of the reninangiotensin-aldosterone hormonal axis. Renin is released from the juxtaglomerular cells of the kidney as a response to diminished renal perfusion, diminished sodium supply to the distal tubule, and beta adrenergic stimulation. Renin converts renin substrate (produced in the liver) into angiotensin I, which is converted mostly in the lungs into angiotensin II, a potent arteriolar vasoconstrictor and a stimulus for aldosterone release from the adrenal glands. Aldosterone production is also augmented by hyperkalemia, adrenocorticotropic hormone (ACTH) elevation, and hyponatremia (
7).
To maintain electroneutrality in the tubular lumen, potassium and hydrogen ions are exchanged for sodium. Hydrogen ion exchange results in increased intracellular bicarbonate. The secreted hydrogen ions react with luminal bicarbonate and reduce bicarbonate concentration in the lumen. Both intracellular and intraluminal reactions of hydrogen ions and bicarbonate are facilitated by carbonic anhydrase. In the distal tubule, hydrogen ion secretion continues, especially when sodium absorption is high (aldosterone secretion). To preserve anion electroneutrality, either chloride or bicarbonate must accompany sodium. During states of chloride deficiency or strong sodium reabsorption (e.g., hypoperfusion of the kidneys, loss of chloride from the stomach, elevated aldosterone or cortisol levels, diuretics), sodium-hydrogen exchange is augmented and results in increased bicarbonate absorption (or intracellular production) and excretion of hydrogen in the face of a metabolic alkalosis (paradoxical aciduria) (
2,
8,
9).
ANP is a potent inhibitor of sodium reabsorption that is released from the atria in response to an increase in atrial stretch. Urodilatin is a pro-ANP fragment that is produced in the kidney and is increased during volume expansion. B-type natriuretic peptide (BNP—previously termed brain natriuretic factor) is principally released from the cardiac ventricles, and some from the brain. BNP concentrations can exceed ANP blood levels during heart failure and in various other critical illnesses (
10,
11).
Water Reabsorption
Water reabsorption is an osmotically driven, passive process that follows sodium migration from the tubular lumen to the interstitial space. However, in the medullary interstitium, sodium is absorbed without water in the ascending loop of Henle. The shape of the loop allows for a progressive increase in interstitial osmolality near the tip of the loop (counter-current mechanism) that can be augmented by vasopressin-induced urea absorption.
In the collecting duct, vasopressin increases water absorption via the V2 receptor that increases the action of pre-formed water channels called aquaporin-2. Water migration through these channels can result in a urine osmolality approaching 1,200 mOsm, the medullary concentration produced by the counter-current mechanism (
3).
Vasopressin secretion is primarily regulated by serum osmolality and the adequacy of systemic perfusion. Inadequate perfusion is a more potent stimulus than osmolar regulation such that hyponatremia can develop as a consequence of circulatory deficits (
12).
Monitoring of Renal Function
The common clinical parameters used to monitor renal function are rate of urine flow and blood urea nitrogen (BUN) and serum creatinine (CR). Too little urine output (oliguria) for the adult is defined as <0.5 cm3/kg/hr. An increase in BUN and/or CR serves to indicate decreasing glomerular filtration.
Acute Kidney Injury
AKI is defined by the criteria listed in
Table 7.3. AKI, like shorter durations of oliguria, can be secondary to pre-renal, intrinsic renal, or post-renal etiologies (
Table 7.4). For hospitalized patients who develop kidney malfunction, the most common intrinsic renal insult is cataloged under the term acute tubular necrosis (ATN), even when anatomical tubular injury is minimal (
vide infra), and the most common post-renal cause is a blocked urinary catheter (rare).
Laboratory Investigation
Urine Osmolality and Specific Gravity
Low urine output secondary to renal hypoperfusion is the normal physiological response to decreased glomerular perfusion and augmented sodium and water reabsorption. Secretion of vasopressin allows tubular reabsorption of water that will increase urine osmolality above plasma levels (i.e., >290 mOsm/L) and results in specific gravity (SG) greater than that of plasma (>1.010, but usually <1.030). Unfortunately, osmolality and SG can be increased by other mechanisms, especially the excretion of osmotically active substances like glucose and and intravenous and/or arterial contrast, which not only increase SG but often promote diuresis. Diuresis with increased SG (especially when >1.030) is a clue that a large, osmotically active molecule is in the urine. A physiologic diuresis secondary to well maintained intravascular volume would more likely decrease SG. After such stress as surgery, trauma, or severe infection, the most likely cause of a large urine output with elevated SG is excretion of glucose secondary
to increased blood glucose from the metabolic response to tissue injury. A urine sugar level will rapidly determine if such is the case.
Urine Electrolytes and Creatinine
With oliguria secondary to hypoperfusion, the augmented sodium reabsorption mechanisms typically result in urine sodium concentrations <20 Meq/L. When urine sodium is >40 Meq/L, the inference is that the renal tubules are injured and cannot respond to aldosterone. The low urine sodium of aldosterone effect is usually associated with a higher urine potassium (>30 Meq/L) even when hypokalemia is present.
Osmotic substances in the urine that result in retention of intraluminal water can limit the magnitude of sodium reabsorption. Usually, however, such a potential effect can be elucidated by measuring the SG. As stated above, a SG >1.030 is suggestive of a non-physiologic osmotic effect on urine chemistries.
The fractional excretion of sodium (FENa), the percent of filtered sodium that is excreted, is considered a more accurate indication of renal hypoperfusion versus poorly functioning tubules.
Values <1.0% are consistent with hypoperfusion; values >2% are most consistent with tubular malfunction. However, several reports have demonstrated FE
Na values <1% in patients with a clinical course more consistent with tubular damage. This is seen most often in patients with intense stimuli for sodium reabsorption (e.g., congestive heart failure, cirrhosis, burns) or particular etiologies of renal damage (e.g., acute glomerular and interstitial nephritis) (
4,
13). In addition, confounders such as loop diuretics and osmotic substances in the urine might increase urine sodium concentrations and the calculated FE
Na despite the presence of a pre-renal state.
BUN and Serum Creatinine
BUN and creatinine levels are easily obtained estimates of glomerular filtration. Urea is a product of liver protein metabolism and can be influenced by the amount of protein administered or the metabolic capability of the liver. A diet deficient in protein coupled with a damaged liver may result in a low BUN despite reduced glomerular filtration. High protein intake with good liver function might produce the opposite effect. Creatinine is the end product of creatinine metabolism in muscle and is relatively constant from day to day. Except with little (e.g., older, wasted patient) or excessive muscle breakdown (rhabdomyolysis), creatinine ranges from 0.6 to 1.0 mg/100 ml in females, and 0.8 to 1.3 mg/100 ml in males with normal glomerular filtration (
3).
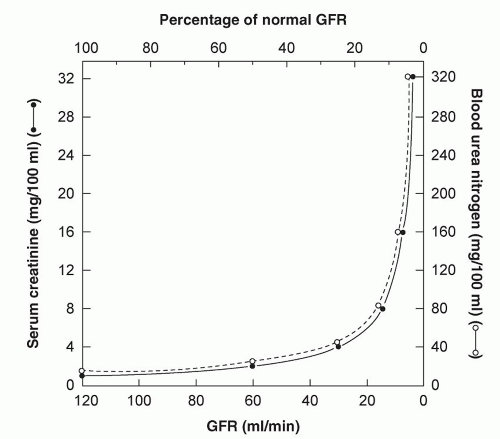
For every 50% reduction in glomerular filtration, serum creatinine doubles (
Fig. 7.1). Therefore, when normal function precedes an insult small increments in creatinine represent relatively large reductions in glomerular filtration. Subsequently, similar increments in creatinine do not indicate the same percentage loss of function. For instance an increase in creatinine from 1.0 to 2.0 mg/dl represents a larger percentage decrease in glomerular filtration than a subsequent increase from 2.0 to 3.0 mg/dl (
14).
After filtration, BUN is also reabsorbed. In oliguric states with preserved tubular function (hypoperfusion), BUN will typically increase more than creatinine and elevate the normal BUN/CR ratio of 10-15 to 20-50 range. Therefore, calculation of the BUN/CR ratio can assist the recognition of hypoperfusion/pre-renal states (
4).
Urinalysis
Urinalysis is particularly useful for determining SG, the presence of glucose, and for evidence of tubular damage (tubular casts) as well as acute glomerular or interstitial alterations (protein, red cell casts, eosinophils) (
4).
Epidemiology of AKI
Reports of an AKI frequency of 20% in hospitalized patients and more than 50% in intensive care unit populations attest to how regularly the kidneys suffer during acute illness (
20). Regardless of etiology, the occurrence of AKI during hospitalization is associated with increased mortality, even when the creatinine increase is as low as 0.3 mg/dl. As might be expected, mortality risk increases with further creatinine elevation and especially when renal replacement therapy is necessary (
Table 7.3) (
4,
21,
22,
23,
24,
25,
26,
27).
Hemodynamic Management of AKI
Pre-renal oliguria is managed by reversing global hypoperfusion. This requires determining the specific etiology of hypoperfusion and providing appropriate therapy (see
chap. 3). The epidemiologic data that link and increase in creatinine to higher mortality do not distinguish between creatinine elevation that is a result of disease versus a result of provider management. Therefore, it is possible that provider-induced renal insult (e.g., depletion of plasma volume with diuresis, administration of renal toxins, inadequate fluid administration) is as detrimental as disease-induced alterations.
For surgical patients, hemodynamic “optimization” using cardiac output measurement and monitors of oxygen utilization is associated with reduced AKI, even when this effort is provided in the post-operative time frame (
28). This finding infers a direct link between provider attention to circulation deficits and the threat of kidney injury. Goal-directed hemodynamic therapy for severe systemic inflammation is also associated with improved renal function (
19).
Once intrinsic renal damage is in place, it is difficult to correlate specific deficits in renal function with renal blood flow, per se (
20). That is, the creatinine might continue to increase despite excellent renal blood flow. However, it is also difficult to presume that injured kidneys benefit from new and/or additional episodes of hypoperfusion. In fact, increasing renal perfusion, usually by attention to global perfusion deficits, may result in more urine output and avoidance of renal replacement, despite further creatinine elevation.
Therefore, the identification and management of inadequate cardiac output is a principal therapeutic concept for renal preservation, just as it is a principal concept for improving patient survival. Since urine output from intrinsically injured kidneys will no longer serve as a practical monitor of global circulation, precise hemodynamic analysis will often demand the use of echocardiograms and invasive monitoring. Patients with AKI should not be maintained in the Ebb phase of shock because of the fear of the effects of resuscitation. Neither the kidneys nor the patient will survive unless the Flow phase is sought and achieved.
Finally, the avoidance of drugs that can alter glomerular blood flow (non-steroidal antiinflammatory agents/analgesics, angiotensin-converting-enzyme inhibitors) is an adjunctive strategy (
29).
Management of Inflammation in AKI
The management of systemic inflammation that can cause AKI is described in the chapter 4. At present, there are no renal-specific anti-inflammatory management strategies, save for attention to the type of dialysis membrane used when hemodialysis is used for renal replacement in severe AKI. The use of a “biocompatible” dialysis membrane that presumably causes less complement activation is associated with improved renal recovery (
30). Inflammation is a principal mechanism for nephrotoxin renal injury. Avoidance of renal toxins is a fundamental strategy in AKI unless toxin administration is unavoidable for optimum management of the patient (e.g., intravenous contrast for suspected pulmonary embolism) (
4).
Rhabdomyolysis results in a toxic injury to the kidneys that is also associated with systemic inflammation that is either a consequence of the muscle injury itself (e.g., crush injury to an extremity) or secondary to another inflammatory process (e.g., pancreatitis). Myoglobin can cause both ischemic renal insults and tubular obstruction. Ischemia-reperfusion-induced
inflammation can then follow the initial ischemic process (
31,
32). While the infusion of bicarbonate and the osmotic diuretic mannitol have been recommended to protect renal tissue from myoglobin toxicity, there is little data supporting the effect of this management strategy. As with other etiologies of AKI, an excellent cardiac output is the most effective therapeutic goal (
31).
Additional Management Concepts for AKI
Utilization of loop diuretics does not result in improved recovery of renal function, although it may result in less need for dialysis. One study concluded that loop diuretics were associated with increased mortality and non-recovery of renal function (
33,
34).
Which fluid to use for plasma volume expansion has been studied, suggesting that crystalloid as compared to synthetic colloids is associated with less frequent AKI (
35).
Cardiac surgery patients who develop AKI appear to respond favorably to the infusion of recombinant human atrial natriuretic peptide (hANP) with less need for renal replacement (
36). The perioperative use of hANP in patients undergoing abdominal aortic aneurysm repair resulted in improved renal function (
37).
The infusion of alkaline phosphatase and the resultant inhibition of nitric oxide production in the kidneys have been shown to improve sepsis-associated AKI in humans (
38).
Renal Replacement Techniques: Continuous Hemofiltration and Dialysis
The relative indications for renal replacement in AKI are listed in
Table 7.6 and the methods used in
Table 7.7. As emphasized in the chapters 3 and 4, total body sodium and water excess does not guarantee increased intravascular volume. However, when truly excessive, increased vascular volume may be removed with renal replacement techniques (especially continuous methods) and potentially improve cardiac function. In such patients, renal replacement may also assist nutritional therapy, allowing several liters of intravenous or intestinal food to be administered. Metabolic acidosis and hyperkalemia are effectively treated by renal replacement, but phosphate filtration is less effective with dialysis as compared to continous techniques. “Uremia” is an ill-defined syndrome that may include altered mental status, pleural/pericardial effusions and coagulopathy. Uremia is associated with blood urea levels close to 100 mg/dl. Certain toxins (antibiotics, salicylates) may be removed only by renal replacement.
Hemodialysis (usually over 3-4 hours) is the most common method used in chronic renal failure and is used frequently in AKI. Hemodialysis in surgical critical illness is often accompanied by hemodynamic instability resulting in administration of fluids and/or drugs to maintain cardiac output. In addition, worsening hypoxemia may be noted. The etiology of these alterations is unclear, but may relate to pulmonary and/or systemic vasodilation in the setting
of normal or low intravascular volume. In one report, daily hemodialysis that removed about two-thirds less ultrafiltration volume as compared to every-other-day hemodialysis was associated with less frequent hypotension, better control of uremia, more recovery of renal function, and lower mortality (
39).
The development of biocompatible “high flux” synthetic membranes with greater permeability than conventional hemodialysis membranes made the development of continuous renal replacement therapy (CRRT) possible in the late 1970s. The addition of a pump into the circuit allowed continuous veno-venous hemodiafiltration (CVVH) and permitted the application of hemofiltration in patients with a narrow arteriovenous pressure gradient.
Peritoneal dialysis via insertion of a catheter into the abdominal cavity is a slower, continuous method of dialysis that does not require vascular access and results in less hemodynamic variation. This approach is rarely used in surgical critical illness.
Solute clearance using RRT may be either convective (filtration) or diffusive (dialysis). If ultrafiltration (CVVH) is used, excess fluid and electrolyte losses must be replaced. Fluid flux may be impressive: more than a liter per hour is not unusual. Sodium, bicarbonate, calcium, phosphate, and magnesium replacement require special attention and all electrolytes require careful monitoring (
40). In addition, therapeutic drugs can be cleared by ultrafiltration often demanding measurement of blood levels and collaboration with a pharmacist to ensure effective concentrations.
When dialysis is the predominant form of solute clearance, the composition of the dialy-sate can make electrolyte balance easier to control. The semipermeable dialysis membrane permits diffusion of solute in both directions. A steady state is eventually approached. Transmembrane pressure differences drive fluid movement but solute clearance depends on the size of the pores in the filter membrane. Molecules smaller than 3,000 Dalton pass readily and drugs of this size are filtered, including vasoactive amines and antibiotics. Again, close pharmacokinetic monitoring is necessary to ensure proper therapeutic dosing. (
41).
Only gold members can continue reading.
Log In or
Register to continue
Related

Full access? Get Clinical Tree