The following items may suggest a possible haemostatic disorder
No
Yes
Situation never encountered
1. Have you previously consulted a doctor or received a treatment for prolonged or unusual bleeding, for example, nosebleeds or small cuts?
2. Do you have a tendency to develop bruises larger than 2 cm or large haematomas in the absence of a bump or wound or else after a minor bump or wound?
3. Have you had to go back to your dentist because of bleeding after a tooth extraction?
4. Have you experienced major bleeding after surgery, for example, adenoidectomy or tonsillectomy, or after circumcision?
5. Do any of your close family members have a coagulation disorder that causes major bleeding, such as von Willebrand disease or haemophilia?
6. For women:
(a) Have you consulted a doctor or received a treatment for heavy menstrual bleeding (e.g. oral contraceptives (“pill”), iron treatment, medication to thicken the blood such as Exacyl)?
(b) Did you experience abnormal bleeding after childbirth?
Score calculated by number of Yes answers to these six questions
17.2 Perioperative Management of Inherited Bleeding Disorders
Some of the rules for perioperative management are common to all inherited bleeding disorders:
Preoperative management of congenital bleeding disorders requires a close multidisciplinary approach involving anaesthesiologists, haematologists, surgeons and the patient’s specialised care centre.
Surgery should take place in a specialised centre or in collaboration with such centre and should be scheduled early in the day on weekdays [3].
Preoperative use of antifibrinolytic agents such as tranexamic acid is an adjuvant therapy of choice [3] and can be given as a 1 g intravenous bolus shortly before anaesthesia induction or started orally (1 g 3–4 times a day) 24–48 h before surgery.
Aspirin and NSAIDs should be avoided for postoperative analgesia, particularly in patients with primary haemostatic disorders.
When levels of the deficient factor are maintained at normal levels, antithrombotic prophylaxis can be considered in patients at risk of thromboembolism.
Inherited bleeding disorders are commonly classified into two main categories: disorders of primary haemostasis and inherited coagulation defects.
17.2.1 Perioperative Management of Congenital Primary Haemostatic Disorders
17.2.1.1 Perioperative Management of von Willebrand Disease
von Willebrand disease (vWD), an autosomal dominant disorder, is the most common inherited bleeding disorder with a prevalence of 1%. It is caused by a quantitative or qualitative deficiency of von Willebrand factor (vWF), which mediates platelet adhesion to the damaged vascular wall and serves as carrier protein for factor VIII (FVIII) protecting it from rapid plasma proteolysis.
Diagnosis is based on plasma assays of vWF (vWF:Ag), its functional activity (ristocetin cofactor activity (vWF:RCo)) and FVIII (FVIII:C).
Depending on whether the deficiency is proportional or not, vWD can be classified into three main groups [4]:
Type 1: partial quantitative deficiency of vWF. This is the most common form.
Type 2: qualitative defects in vWF are divided into four subtypes: 2A (decreased affinity for platelet GPIb associated with the absence of high molecular weight vWF multimers), 2B (increased affinity), 2M (decreased vWF affinity for platelet GPIb with the presence of all multimers) and 2N (decreased affinity for FVIII).
Type 3: complete absence of vWF. This is the most severe form.
vWF:RCo and FVIII levels should be assayed in the week before surgery and the absence of vWF inhibitor should be ascertained (type 3).
Principles of Perioperative Replacement Therapy
There are two treatments of choice for vWD and the indication depends on the type: desmopressin (1-deamino-8-d-arginine vasopressin, also called DDAVP) and infusion of vWF/FVIII concentrates.
In responders, DDAVP (Minirin®) induces a two- to fourfold increase in FVIII and vWF levels 30–60 min after infusion via release of endogenous vWF stored in endothelial cells, with a return to baseline occurring after 6–9 h [5]. This is the treatment of choice for type 1 patients with baseline vWF level >10 IU/dL. It can also be used in some patients with the 2A, 2M or 2N subtype [4]. The response to DDAVP should be assessed prior to surgery because there is large interindividual variability. In particular, some von Willebrand subtypes exhibit increased clearance of vWF and would require more closely spaced infusions [4]. In type 2B, DDAVP is contraindicated because of the risk of appearance or aggravation of thrombocytopenia.
In responders, DDAVP should be administered subcutaneously or intravenously 30–60 min before surgery (Fig. 17.1). Repeat doses can then be given every 12–24 h. Tachyphylaxis and hyponatraemia may occur, which is why fluid intake should be limited and serum sodium monitored in case of repeated doses [5].
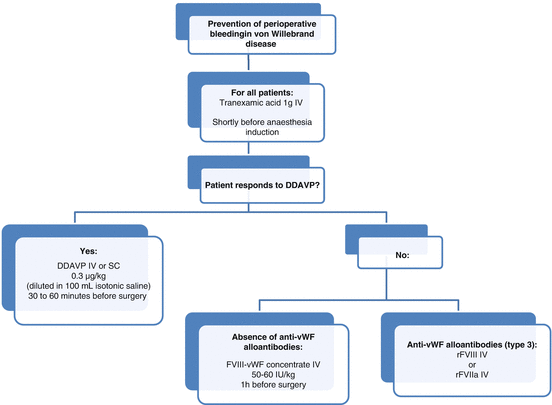
Fig. 17.1
Algorithm for surgical management of patients with von Willebrand disease. DDAVP desmopressin, IV intravenous, SC subcutaneous
When desmopressin is contraindicated or ineffective, replacement therapy with either vWF alone or in combination with FVIII is used. Several vWF/FVIII products are available on the market, each having a different vWF:RCo/FVIII:C ratio. These combined products simultaneously increase vWF and FVIII levels, in contrast to vWF concentrates where there is a delay of 6–12 h before endogenous FVIII begins to increase. A loading dose of FVIII-vWF concentrate is recommended 1 h before surgery (Fig. 17.1).
Furthermore, in December 2015 the US Food and Drug Administration (FDA) approved the first recombinant vWF (Vonvendi®, Baxalta, USA) for the treatment of bleeding events in von Willebrand patients [6].
There are not yet enough validated data to clearly define the haemostatic target levels of vWF and FVIII and the duration of postoperative replacement therapy (Table 17.2).
Table 17.2
Recommended haemostatic target levels for Willebrand ristocetin cofactor (vWF RCo)/factor VIII (FVIII) according to type of surgery
Invasive intervention | Days | Haemostatic target levels |
|---|---|---|
vWF RCo/FVIII (%) | ||
Major surgery | D0 | 100 |
D1–D5/10 | ≥50 | |
Minor surgery | D0 | ≥50 |
D1–D2/D4 | ≥30 |
vWF:RCo and FVIII levels should be measured daily during the postoperative period. When FVIII level rises above 50%, replacement therapy should be continued with a vWF concentrate alone, so as to decrease the risk of venous thrombosis [4].
Special Cases
von Willebrand Disease with Inhibitors
Pregnancy and von Willebrand Disease
The physiological two- to threefold increase in vWF and FVIII levels that occurs during pregnancy generally suffices to correct the vWF deficiency in patients with type 1 but not type 2 or 3. Several learned societies have issued recommendations for the peripartum management of these patients, and they all agree that Caesarean section, vaginal delivery and neuraxial anaesthesia can be performed safely when vWF:RCo and FVIII levels are ≥50% during the third trimester [7]. On the other hand, in patients with vWF/FVIII ≤50%, who generally have type 2 or 3, there are no clear guidelines on the target levels to be achieved by replacement therapy at the time of delivery nor on the optimal duration of postpartum replacement therapy. Some authors recommend that vWF:RCo/FVIII haemostatic target levels should be between 150 and 200% shortly before delivery and be maintained at these levels for the first 4–7 days postpartum [8]. Furthermore, neuraxial anaesthesia is contraindicated in types 2 and 3 [7].
17.2.1.2 Perioperative Management of Inherited Platelet Disorders
The inherited platelet disorders are characterised by abnormal expression of platelet receptors or granule secretion, affecting one or more steps of platelet activation [9].
Among the most severe inherited platelet function disorders, two are of particular interest: Glanzmann thrombasthenia, caused by quantitative or qualitative abnormalities in the platelet fibrinogen receptor, glycoprotein GPIIb-IIIa, and Bernard-Soulier syndrome, characterised by defective platelet adhesion to vascular subendothelium linked to an abnormality in the GPIb-IX-V receptor complex.
Multiple platelet transfusions may induce the formation of antibodies directed against the HLA system and/or against glycoproteins GPIIb-IIIa in patients with Glanzmann thrombasthenia; patients should be screened for the presence of such antibodies in the week before surgery.
For minor surgery and mild forms of platelet disorders, local haemostatics, together with systemic tranexamic acid and, in some cases, with DDAVP, are usually sufficient in most cases. DDAVP is not effective in platelet disorders characterised by platelet effector receptor defects (Fig. 17.2) [9].
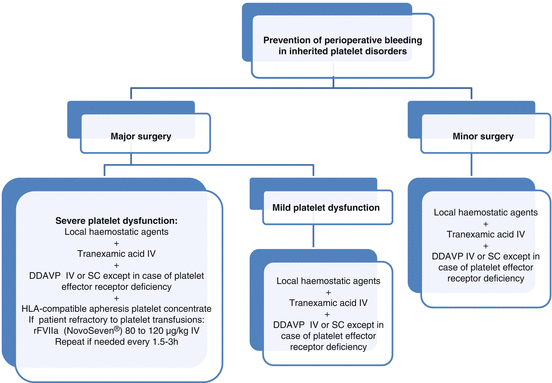
Fig. 17.2
Algorithm for surgical management of inherited platelet disorders. DDAVP desmopressine, IV intravenous, SC subcutaneous
For major surgery in patients with moderate to severe platelet disorders, prophylaxis is based on transfusion of HLA-matched apheresis platelets together with the adjuvant treatments noted above (Fig. 17.2) [3, 9].
The presence of antibodies is not always a predictor of refractoriness to platelet transfusion and should not by itself be a contraindication to platelet transfusion. Recombinant FVIIa (NovoSeven®) is another alternative in immunised or nonimmunised patients refractory to platelet transfusions (Fig. 17.2) [3].
Postoperatively, platelet transfusions or rFVIIa can be administered for variable lengths of time, sometimes until wound healing.
It should also be noted that spinal anaesthesia is contraindicated.
17.2.2 Perioperative Management of Inherited Coagulation Disorders
17.2.2.1 Perioperative Management of Haemophilia
Haemophilia is a recessive X-linked disorder caused by a deficiency of factor VIII (FVIII) (haemophilia A, accounting for 80% of haemophilia) or factor IX (FIX) (haemophilia B).
The severity of haemophilia is arbitrarily divided into three groups: severe when FVIII or FIX <1%, moderate 1–5%, and mild >5%.
Diagnosis is suspected in the case of a prolonged aPTT (activated partial thromboplastin time) and confirmed by assay of FVIII and FIX.
In the week before surgery, it is essential to measure FVIII and IX levels and to screen for FVIII and FIX inhibitors [3].
Principles of Perioperative Replacement Therapy
The deficient factor should be administered shortly before induction of anaesthesia in order to avoid a too early decline in plasma levels of the factor during surgery.
FVIII concentrates are the treatment of choice in haemophilia A (Fig. 17.3). In the absence of inhibitors, each unit of FVIII administered per kg of body weight increases the plasma level by 2 IU/dL [10]. FVIII should be administered by slow intravenous injection not exceeding 3 mL/min. The half-life is 8–12 h.
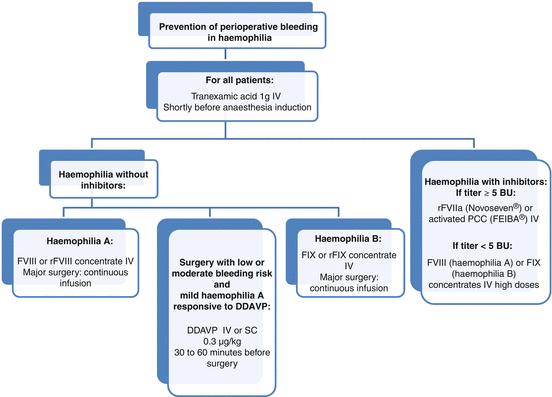
Fig. 17.3
Algorithm for surgical management of patients with haemophilia. DDAVP desmopressine, IV intravenous, SC subcutaneous
For FIX deficiency, it is preferable to use a product that contains only FIX instead of prothrombin complex concentrates (PCC) which contain other coagulation factors that could increase the risk of thrombosis (Fig. 17.3). In the absence of inhibitors, each unit of plasma-derived or recombinant FIX administered per kg of body weight increases the level of FIX by 1 IU/dL and 0.8 IU/dL, respectively [10]. The half-life of FIX is 18–24 h.
European guidelines for the haemostatic target levels of the deficient factor and the duration of postoperative replacement according to type of surgery are shown in Table 17.3.
Table 17.3
Recommended haemostatic target levels for factor VIII (FVIII) and factor IX (FIX) according to type of surgery
Invasive intervention | Days | Haemostatic target level |
|---|---|---|
FVIII/FIX (%) | ||
Major surgery | D0 | ≥80 |
D1–D7 | ≥50 | |
D8–D21 | ≥30 | |
Minor surgery | D0–D4 | ≥50 |
D5–D6/D8 | ≥20 |
In the case of major surgery, continuous infusion should be preferred because it produces more stable factor levels and reduces factor consumption as compared to discontinuous administration [12].
It is generally not necessary to measure the factor levels during surgery, but postoperatively, FVIII and FIX should be assayed daily. Unlike FVIII, FIX levels do not rise in response to inflammation, such that longer postoperative replacement therapy is usually necessary.
Special Cases
Mild Haemophilia A
Haemophilia with Inhibitors
In patients who develop inhibitors, perioperative management is usually based on administration of rFVIIa or activated PCC (FEIBA factor eight inhibitor bypassing agent®, Baxter Healthcare Corp, Thetford, Norfolk, UK) (Fig. 17.3) [3]. The purpose of these agents is to increase the formation of thrombin, a key enzyme that cleaves fibrinogen to fibrin, by bypassing the intrinsic tenase complex formed by activated FIXa and FVIIIa.
Related posts:





Full access? Get Clinical Tree


