The Liver: Surgery and Anesthesia
Randolph H. Steadman
Michelle Y. Braunfeld
Key Points
Related Matter
Left Liver Lobe Donation
Hepatic Function in Health
The liver is the largest internal organ and is the body’s metabolic headquarters. It weighs 1.5 kg or about 2% of the total body weight in an adult. The functional unit of the liver is the lobule, a structure roughly 1 × 2 mm that consists of plates of hepatocytes located in a radial distribution about a central vein. The afferent blood supply from the portal vein and hepatic arteriole enters at the periphery of the lobule. Bile, formed in the hepatocytes, flows into canaliculi located between the plates of hepatocytes and drains into bile ducts located at the periphery of the lobule next to portal venules and hepatic arterioles. The large pores in the endothelium lining the sinusoids allow plasma and its proteins to move readily into the tissue spaces surrounding hepatocytes, an area known as the spaces of Disse. This fluid drains into the lymphatic system. The liver generates about half of the body’s lymph (Fig. 45-1).
The liver’s high blood flow is due to low vascular resistance in the portal vein. The average portal vein pressure is 8 to 10 mm Hg while the hepatic venous pressure averages 0 mm Hg. However, when injured hepatocytes are replaced by fibrous tissue, blood flow is impeded and portal hypertension ensues. Sinusoidal pressures greater than 5 mm Hg are abnormal and define portal hypertension.1 Sympathetic innervation from T3 to T11 controls resistance in the hepatic venules. Changes in compliance in the hepatic venous system contribute to the regulation of cardiac output and blood volume. In the presence of reduced portal venous flow, the hepatic artery can increase flow by as much as 100% to maintain hepatic oxygen delivery. The reciprocal relationship between flow in the two afferent vessels is termed the “hepatic arterial buffer response.”2
The microcirculation of the liver lobule is divided into three zones that receive varying oxygen content.3 Zone 1 receives oxygen-rich blood from the adjacent portal vein and hepatic artery. As blood moves through the sinusoid it passes from the intermediate zone 2 into zone 3, which surrounds the central vein. Blood entering zone 3 is oxygen poor. Pericentral hepatocytes have a greater quantity of cytochrome P450 enzymes and are the site of anaerobic metabolism. Hypoxia and reactive metabolic intermediates from biotransformation affect zone 3 more prominently than other zones.
Due to its ability to distend, the liver is capable of storing up to 1 L of blood. The liver serves as a reservoir capable of accepting blood, or releasing blood at times of low blood volume. The liver also stores vitamins, particularly vitamins B12 (1-year supply), D (3-month supply), and A (10-month supply). Excess body iron is transported via apoferritin to the liver for storage as ferritin, which is released when circulating iron levels are low. Thus, the liver apoferritin system serves as a blood iron buffer.
Reticuloendothelial cells called Kupffer cells line the venous sinusoids. These macrophages phagocytize bacteria that enter the sinusoids from the intestines. Less than 1% of bacteria that enter the liver pass through the systemic circulation.
The liver synthesizes fat, cholesterol, phospholipids, and lipoproteins. It also efficiently metabolizes fat, converting fatty acids to acetyl coenzyme A (Co A), an excellent source of energy, which can be diverted to the citric acid cycle to liberate energy for the
liver. The liver generates more acetyl-CoA than it consumes. The excess is packaged as acetoacetic acid for use elsewhere in the body. The majority of cholesterol synthesized in the liver is converted to bile salts and secreted in the bile. The remainder is distributed to the rest of the body where it is used to form cellular membranes and other vital structures. Fat synthesis from protein and carbohydrates occurs almost exclusively in the liver, and the liver is responsible for most fat metabolism.
liver. The liver generates more acetyl-CoA than it consumes. The excess is packaged as acetoacetic acid for use elsewhere in the body. The majority of cholesterol synthesized in the liver is converted to bile salts and secreted in the bile. The remainder is distributed to the rest of the body where it is used to form cellular membranes and other vital structures. Fat synthesis from protein and carbohydrates occurs almost exclusively in the liver, and the liver is responsible for most fat metabolism.
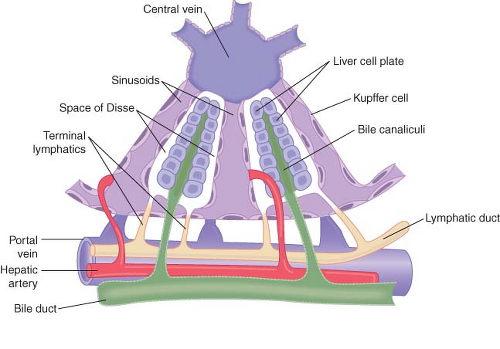 Figure 45-1. Basic structure of a liver lobule, showing the cellular plates, the blood vessels, the bile-collecting system, and the lymph flow system composed of the spaces of Disse and the interlobular lymphatics. (Modified from: Guyton AC, Taylor AE, Granger HJ. Circulatory Physiology. Vol 2. Dynamics and Control of the Body Fluids. Philadelphia, PA: WB Saunders; 1975.) |
The liver also plays a key role in protein metabolism. The liver synthesizes all of the plasma proteins with the exception of γ-globulins, which are formed in plasma cells. The liver is capable of forming 15 to 50 g of protein per day, an amount sufficient to replace the body’s entire supply of protein in several weeks. Albumin is the major protein synthesized by the liver and is the primary determinant of plasma oncotic pressure. The liver also synthesizes the nonessential amino acids from keto acids, which are also synthesized in the liver.
The liver is capable of deamination of amino acids, which is required for energy production or the conversion of amino acids to carbohydrates or fats. Deamination produces ammonia, which is toxic. Intestinal bacteria are an additional source of ammonia. The liver removes ammonia through the formation of urea.
All of the blood clotting factors, with the exception of factors III (tissue thromboplastin), IV (calcium), and VIII (von Willebrand factor), are synthesized in the liver. Vitamin K is required for the synthesis of prothrombin (factor II) and factors VII, IX, and X.
Hepatocytes produce roughly 500 mL of bile daily. Between meals, the high pressure in the sphincter of Oddi diverts bile to the gallbladder for storage. The gallbladder holds 35 to 50 mL of bile in concentrated form. The presence of fat in the duodenum causes release of the hormone cholecystokinin from duodenal mucosa, which reaches the gallbladder via circulation and stimulates gallbladder contraction. Bile contains bile salts, bilirubin, and cholesterol. Bile salts act as a detergent, solubilizing fat into micelles, which are absorbed. Bile salts return to the liver via the portal vein, completing the enterohepatic circulation. Bile salts are needed for fat absorption, and cholestasis can result in steatorrhea and vitamin K deficiency.
The liver has the unique ability to restore itself after injury or partial hepatectomy. As much as two-thirds of the liver can be removed with regeneration of the remaining liver in a matter of weeks.5 Hepatocyte growth factor, produced by mesenchymal cells in the liver, and other growth factors, such as epidermal growth factor (EGF), cytokines, tumor necrosis factor (TNF), and interleukin-6, are involved in stimulating regeneration. Growth factor-β, a known inhibitor of hepatocyte proliferation, is involved in halting the regenerative process, which appears to be related to the ratio of liver-to-body weight.5,6 Inflammation, such as with a viral infection of the liver, impairs regeneration.
Assessment of Hepatic Function
A number of laboratory tests are available to assess the liver. Collectively termed liver function tests (LFTs) many, including aspartate aminotransferase (AST) and alanine aminotransferase (ALT), do not assess function but rather cellular injury. Increased serum levels of these enzymes, AST (formerly serum glutamic oxaloacetic transaminase or SGOT) and ALT (formerly serum glutamic pyruvic transaminase or SGPT), occur in many types of hepatic disease. Because AST is also found in nonhepatic tissues (including the heart, skeletal muscle, kidney, and brain), elevations are not specific for hepatic disease. ALT is primarily localized to the liver.
Fatty liver and chronic infections are associated with mild (several fold) elevations of AST and ALT. Acute hepatitis produces larger increases, but the highest concentrations, which can exceed 50 times normal, are seen with acute hepatitis necrosis. Absolute levels of these enzymes are not always helpful, as declining values may indicate recovery or conversely a lack of surviving hepatocytes. The AST/ALT ratio may be helpful in differentiating alcoholic liver disease, in which the ratio is typically >2, from viral hepatitis, which is associated with a ratio <1.
Indices of bile flow obstruction include serum levels of alkaline phosphatase (AP), 5′-nucleotidase (5′-NT), γ-glutamyl transferase (GGT), and bilirubin. AP isoenzymes are found in multiple organs including the liver, bone, kidney, intestines, placenta, and leukocytes. Normally, most circulating AP originates from liver and bone. Hepatic AP is concentrated in the microvilli of bile canaliculi and the sinusoidal surface of hepatocytes. Elevations of serum AP disproportionate to changes in AST and ALT occur with obstructions to bile flow. However, AP elevations may originate from other tissues, including the placenta during pregnancy. Although 5′-NT is also found in many tissues, elevations are highly specific for hepatobiliary obstruction. Elevations of 5′-NT may reflect the detergent action of bile salts on plasma membranes, a requirement for its release. Because 5′-NT is so specific for liver diseases, it is useful to determine whether elevated AP is of hepatic origin. Serum GGT is the most sensitive laboratory indicator of biliary tract disease but it is less specific than 5′-NT and has largely been replaced by 5′-NT.
Bilirubin originates primarily from the breakdown of hemoglobin released from senescent red blood cells. Serum bilirubin levels are determined by the van den Bergh reaction, which separates bilirubin into two fractions: A lipid-soluble, indirect-reacting form (unconjugated bilirubin) and a water-soluble, direct-reacting form (conjugated bilirubin). Elevated levels of unconjugated bilirubin indicate an excess production of bilirubin (hemolysis) or a decrease in the uptake and conjugation of bilirubin by hepatocytes. Conjugated bilirubin is elevated by impaired intrahepatic excretion or extrahepatic obstruction. Even with complete biliary tract obstruction, the bilirubin rarely exceeds 35 mg/dL because of renal excretion of conjugated bilirubin.
Tests of hepatic synthetic function focus on the measurement of serum albumin and coagulation testing. Although the liver is the primary site of albumin synthesis, excessive protein losses (enteropathy, burns, nephrotic syndrome) can also result in low albumin levels. Because of its 3-week half-life, serum albumin is not a reliable indicator of acute liver disease. In contrast, the prothrombin time (PT) and international normalized ratio (INR) are sensitive indicators of hepatic disease because of the short half-life of factor VII. The PT depends upon sufficient intake of vitamin K, which in turn depends upon adequate biliary secretion of bile salts. In patients with biliary obstruction, the PT can be prolonged despite preserved hepatic function. Other conditions that can affect the PT in the absence of liver disease include congenital coagulation factor deficiencies, consumptive coagulopathies such as disseminated intravascular coagulation (DIC), and warfarin therapy.
A number of other tests exist to assess hepatic function, though their use in the United States is limited primarily to research applications. Indocyanine green (ICG) elimination estimates hepatic blood flow and hepatocellular function due to the high extraction ratio of ICG (>70%). The MEGX test measures the conversion of lidocaine to monoethylglycinexylidide (MEGX) via hepatic demethylation. Other metabolic tests include antipyrine clearance, aminopyrine breath test, caffeine breath test, galactose elimination capacity, and urea synthesis.
Ancillary tests to confirm specific diagnoses include serologic tests for the various hepatitis viruses, autoantibodies (for
the diagnosis of primary biliary cirrhosis [PBC]), ceruloplasmin (Wilson’s disease), ferritin (hemochromatosis), α-1 antitrypsin (α-1 antitrypsin deficiency), and α-fetoprotein (hepatocellular carcinoma [HCC]). Serum ammonia is useful for following patients with hepatic encephalopathy (HE).
the diagnosis of primary biliary cirrhosis [PBC]), ceruloplasmin (Wilson’s disease), ferritin (hemochromatosis), α-1 antitrypsin (α-1 antitrypsin deficiency), and α-fetoprotein (hepatocellular carcinoma [HCC]). Serum ammonia is useful for following patients with hepatic encephalopathy (HE).
Hepatobiliary Imaging
Selection of the appropriate imaging technique depends on the differential diagnosis and whether a concurrent therapeutic intervention is planned. Plain radiography has a limited role in the evaluation of liver disease. Abdominal X-rays can be useful to detect calcified or gas-containing lesions. Examples include calcified gallstones, chronic calcific pancreatitis, gas-containing liver abscesses, portal venous gas, and emphysematous cholecystitis.
Ultrasonography is the primary screening test for hepatic parenchymal disease and extrahepatic biliary disease. It is the method of choice for detecting gallstones, the presence of ascites, and portal or hepatic vein thrombosis. Its major limitations are its dependence on the operator’s skill and its inability to penetrate bone or air, including bowel gas.
Radioisotope scanning has largely been replaced by computed tomography (CT) scanning. However, it is still in use in patients with suspected acute cholecystitis. Radioisotopes visualized in the gallbladder rule out obstruction of the cystic duct, while visualization of the biliary tree and common bile duct without the gallbladder indicates cystic duct obstruction and the presence of cholecystitis.
CT scanning supplements ultrasonography, providing information on the liver texture, gallbladder disease, bile duct dilatation, and mass lesions of the liver and pancreas. CT provides more resolution than ultrasonography and is less operator-dependent. Lesions can be biopsied under CT guidance. The disadvantages of CT include radiation exposure and cost.
Magnetic resonance imaging (MRI) is increasingly used for the evaluation of hepatobiliary disease. MRI is superior to CT for the assessment of malignant focal liver lesions and diffuse liver disease.7 MRI is also useful for the evaluation of biliary disease.8 MRI also offers the advantages of avoidance of radiation and contrast nephropathy. The primary disadvantage is the need for a 20-second breath-hold, which can require sedation or anesthesia in young and/or uncooperative patients.
Percutaneous transhepatic cholangiography (THC) is the percutaneous injection of contrast into the bile ducts under fluoroscopic guidance. It can be used to determine the site and cause of biliary obstruction and to evaluate whether cholangiocarcinoma is surgically resectable. It can also be used for balloon dilatation of biliary strictures and/or placement of an internal stent or external drain. Endoscopic retrograde cholangiopancreatography (ERCP) uses endoscopy to visualize the ampulla of Vater and selectively inject contrast material into the pancreatic and common bile ducts. ERCP has the advantage over THC of not requiring a dilated biliary tree to achieve a high probability of success. ERCP permits sphincterotomy and stone extraction, biopsy, brushings, balloon dilatation, and stent insertion.
Liver Biopsy
Liver biopsy continues to have a role in the evaluation of patients with liver disease. It is the method of choice to determine whether liver damage is due to necrosis, inflammation, steatosis, or fibrosis. The presence of coagulopathy or thrombocytopenia contraindicates percutaneous liver biopsy, although transjugular liver biopsy can be performed under these conditions.
Hepatic and Hepatobiliary Diseases
Liver disease may be the result of a variety of causes, which include developmental or genetic defects, metabolic abnormalities, autoimmune diseases, infectious diseases, neoplasm, alcohol, environmental toxins, and drug toxicity. A preliminary report from the National Vital Statistics System for the year 2009 lists liver disease as the 12th leading cause of death in the United States, being responsible for over 33,000 deaths in that year. An estimated 5.5 million people in the United States have chronic liver disease (CLD) and another 20 million have biliary disease, together affecting slightly more than 8% of the population. Economic costs for care of these conditions total $8.9 billion per year.9
Liver disease can be divided into two main groups on the basis of the primary anatomy affected. Processes may be considered primarily hepatocellular (parenchymal) or biliary. Progressive biliary disease may eventually lead to fibrotic changes and cirrhosis, but it is characteristic of the biliary diseases that cholestasis precedes hepatocellular dysfunction. In hepatocellular diseases evidence of cholestasis and synthetic dysfunction appear synchronously (Table 45-1). The fact that hepatocellular function is preserved until late in course of cholestatic disease disadvantages patients with cholestatic liver disease awaiting liver transplantation. Priority for transplant is based on the model for end-stage liver disease (MELD) score, which is sensitive to hepatocellular dysfunction. Patients with cholestatic disease may lose transplant candidacy because of other manifestations of their disease, for example, malignancy, infection, or concurrent autoimmune disease, before they accrue enough hepatocellular damage to be offered a transplant.10
Liver disease may also be described as acute or chronic. The most common causes for acute liver disease are drug toxicity and infection. Acute illnesses may resolve spontaneously, segue into chronic disease, or result in acute liver failure (ALF). While the primary cause of ALF in the United States was once infectious (presumed acute hepatitis A and B), acetaminophen toxicity is currently the leading cause of this condition. Other causes of acute liver dysfunction include alcoholic hepatitis, nonacetaminophen drug toxicity, and pregnancy-related hepatic diseases. The most common causes for CLD are chronic viral hepatitis, alcoholic liver disease, and nonalcoholic fatty liver disease (NAFLD). Although the prevalence rates of chronic viral hepatitis and alcoholic liver disease have been relatively stable over the past 10 years, the prevalence of NAFLD has grown significantly and appears to be linked to the current epidemic of obesity.11 The most important consequences of CLD are portal hypertension, cirrhosis, and malignancy.
Acute Liver Failure
ALF (previously termed fulminant hepatic failure) is defined as the appearance of encephalopathy together with coagulopathy, usually an INR ≥1.5, in a patient who has no previous history of liver disease and who has had an illness of <26 weeks’ duration. Although further distinctions in duration of disease, such as hyperacute and subacute, were once used they are no longer
considered useful for prognosis and have been abandoned. ALF is a rare entity with an incidence of about 2,000 cases per year in the United States. Drug-related toxicity accounts for over half of the cases of ALF in the United States. Of these drug-related cases, over 80% are the result of acetaminophen ingestion. In descending order the next most common causes are idiopathic, acute viral hepatitis, autoimmune, and ischemic.12 The natural history of adult ALF in the United States is one of spontaneous recovery in approximately 45% of patients, liver transplantation in 25%, and death without transplantation in 30%.13 Etiology has some bearing on outcome, with the most favorable prognosis for patients with acetaminophen overdose, ischemic injury, and hepatitis A and poor prognoses for those with nonacetaminophen drug-induced liver injury (DILI), acute hepatitis B, Wilson’s disease, and autoimmune hepatitis.14
considered useful for prognosis and have been abandoned. ALF is a rare entity with an incidence of about 2,000 cases per year in the United States. Drug-related toxicity accounts for over half of the cases of ALF in the United States. Of these drug-related cases, over 80% are the result of acetaminophen ingestion. In descending order the next most common causes are idiopathic, acute viral hepatitis, autoimmune, and ischemic.12 The natural history of adult ALF in the United States is one of spontaneous recovery in approximately 45% of patients, liver transplantation in 25%, and death without transplantation in 30%.13 Etiology has some bearing on outcome, with the most favorable prognosis for patients with acetaminophen overdose, ischemic injury, and hepatitis A and poor prognoses for those with nonacetaminophen drug-induced liver injury (DILI), acute hepatitis B, Wilson’s disease, and autoimmune hepatitis.14
Table 45-1. Blood Tests and the Differential Diagnosis of Hepatic Dysfunction | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
Patients with no previous history of liver disease who present with signs or symptoms and laboratory evidence of a significant hepatitis should have an INR measured and undergo a careful mental status examination. An INR ≥1.5 and any evidence of encephalopathy should lead to admission to the hospital for ALF. History should include questions about potential infectious or toxic exposures and a detailed history of recent medications or ingestions. Questions should include details about herbal and nutritional supplements, since these have been associated with ALF as well. Except for the finding of encephalopathy, physical examination may be unrevealing. In particular, evidence of CLD should not be present, as the patient should not have had adequate time to develop the stigmata of portal hypertension and cirrhosis. Acute decompensation of CLD or “acute on chronic” liver disease is a separate condition with different etiologies, therapy, and prognostic indicators.
Standard initial labs are indicated in Table 45-2.14 Further laboratory and investigative studies are directed by the history, for example, radiologic imaging or ultrasound for suspected hepatic vein thrombosis. Although etiologies of ALF are heterogenous, there are manifestations that are common to all patients who have massive hepatic necrosis, regardless of its provenance. The most
serious, and often the proximate cause of death, is acute cerebral edema and intracranial hypertension. Effects on other organ systems include coagulopathy, circulatory dysfunction and hypotension, acute kidney injury, and metabolic derangements. Encephalopathy is a necessary finding to diagnose ALF. Encephalopathy is graded on a I to IV scale and is described in Table 45-3. The presence of cerebral edema is directly related to the depth of encephalopathy. The incidence of cerebral edema is almost negligible in stage I to stage II coma, but increases to 25% to 35% with stage III and 75% with stage IV.15 As with the encephalopathy of cirrhosis, the underlying mechanism is not completely understood but hyperammonemia plays a significant role. Ammonia, which is toxic, is generally metabolized via the urea cycle in the liver. The brain has no cells capable of utilizing the urea cycle and thus must resort to detoxifying ammonia by synthesizing glutamine from ammonia and glutamate within astrocytes. Glutamine is osmotically active and results in osmotic astrocyte edema. Other contributors to the observed cerebral edema may include a systemic inflammatory response16,17 and the loss of cerebral autoregulation, which leads to cerebral hyperemia.18 Potential targets for therapy include osmotic and mechanical reduction of cerebral edema, elimination of ammonia, manipulation of cerebral blood flow and metabolism, and reduction of the inflammatory response.
serious, and often the proximate cause of death, is acute cerebral edema and intracranial hypertension. Effects on other organ systems include coagulopathy, circulatory dysfunction and hypotension, acute kidney injury, and metabolic derangements. Encephalopathy is a necessary finding to diagnose ALF. Encephalopathy is graded on a I to IV scale and is described in Table 45-3. The presence of cerebral edema is directly related to the depth of encephalopathy. The incidence of cerebral edema is almost negligible in stage I to stage II coma, but increases to 25% to 35% with stage III and 75% with stage IV.15 As with the encephalopathy of cirrhosis, the underlying mechanism is not completely understood but hyperammonemia plays a significant role. Ammonia, which is toxic, is generally metabolized via the urea cycle in the liver. The brain has no cells capable of utilizing the urea cycle and thus must resort to detoxifying ammonia by synthesizing glutamine from ammonia and glutamate within astrocytes. Glutamine is osmotically active and results in osmotic astrocyte edema. Other contributors to the observed cerebral edema may include a systemic inflammatory response16,17 and the loss of cerebral autoregulation, which leads to cerebral hyperemia.18 Potential targets for therapy include osmotic and mechanical reduction of cerebral edema, elimination of ammonia, manipulation of cerebral blood flow and metabolism, and reduction of the inflammatory response.
Table 45-2. Initial Laboratory Analysis of Suspected Acute Liver Failure | ||
|---|---|---|
|
Table 45-3. Grades of Encephalopathy | ||
|---|---|---|
|
General measures to reduce cerebral edema include maintaining the patient in a 30-degree head-up position and making sure the head is in neutral position so as not to impede venous return. Once a patient is intubated, muscle relaxants should be considered to minimize rises in intracranial pressure (ICP) from coughing, bucking, and shivering. Mannitol can be used to induce an osmotic diuresis, but may have limited utility in the patient with compromised renal function. Another option may be hypertonic saline, ideally targeting a serum sodium of 145 to 155 mEq/L.19
Although hyperventilation may acutely reduce the cerebral hyperemia associated with ALF, the response is short-lived. There is no evidence that chronic hyperventilation affords any decrease in episodes of intracranial hypertension or any survival benefit.20 Current recommendations are to maintain normocarbia and to reserve hyperventilation for response to acute rises in ICP. Barbiturates can be used to decrease cerebral metabolism; however, their use may be limited by hypotension.
Ammonia can be eliminated by the administration of lactulose or nonabsorbable antibiotics such as rifaximin or neomycin; however, there is no evidence to support their use in the setting of ALF. Furthermore, neomycin is specifically contraindicated because of the risk of nephrotoxicity.
Corticosteroids have not been shown to be effective in ALF, but there may be a place for prophylactic antibiotics to prevent sepsis and minimize the inflammatory mediator burden. The U.S. Acute Liver Failure Study Group has recommended empiric administration of antibiotics in the following settings: (a) when surveillance cultures reveal significant isolates; (b) progression to stage III or stage IV coma; (c) refractory hypotension; or (d) when the patient exhibits elements of the systemic inflammatory response syndrome, that is, temperature >38° or <36°, heart rate >90 bpm, white blood cell count >12,000 or <4,000.21 Other potential modalities to decrease the inflammatory response include modest hypothermia to a target temperature of 32° to 34°C and indomethacin.22
How to monitor the presence and progression of cerebral edema and intracranial hypertension is controversial. Serial head CTs are often obtained for patients who progress to stage III to stage IV coma, but they are not reliable for diagnosing or quantitating intracranial hypertension due to a lack of sensitivity. CT can, however, provide information on structural abnormalities such as intracranial hemorrhage.23 Although many centers will place an ICP monitor to guide therapy in patients with stage III to stage IV coma, there are no randomized controlled studies to support this practice. Furthermore, ICP monitor placement is not a benign procedure, frequently entailing aggressive correction of coagulopathy and transport to and from the OR for a critically ill, fragile patient. Nonetheless, many believe that ICP monitors are invaluable for guiding acute therapy and for helping to determine who may no longer be a viable candidate for transplantation. In addition to measuring ICP, these monitors allow calculation of cerebral perfusion pressure (CPP = MAP – ICP), which should be kept between 50 and 80 mm Hg. In one case series, a sustained CPP <40 mm Hg for greater than 2 hours was associated with a poor neurologic outcome.24 An effective protocol for managing intracranial hypertension in patients with stage III or stage IV encephalopathy has been described (Table 45-4) and resulted in a 95% response to treatment of episodes of ICP >20 mm Hg. Furthermore, in this prospective series, ICP was monitored in all patients, and no patients died of isolated cerebral edema. The authors used a protocol that included activated recombinant factor VII (rFVIIa) to correct coagulopathy prior to ICP placement. Significant bleeding complications from ICP monitoring were not encountered.25
Table 45-4. ICP Management Protocol | ||
|---|---|---|
|
Table 45-5. King’s College Selection Criteria for Liver Transplantation According to the Etiology of Acute Liver Failure | ||||||||
|---|---|---|---|---|---|---|---|---|
| ||||||||
Equally controversial to the monitoring and significance of ICP is determining the prognosis for patients with ALF. Organs for transplantation are a scarce resource, and for some patients liver transplantation is the only life-saving option. However, this life-saving procedure comes with the requirements of major surgery and lifelong immunosuppression. Deciding which patient should receive a transplant, which may recover spontaneously, and which are unlikely to benefit from transplantation is one of the most difficult decisions encountered during the management of patients with liver disease. Unfortunately there is no ideal guideline for making these decisions. The two most widely used prognostic models are the Clichy or Paul Brousse Hospital criteria and the King’s College Hospital criteria. The Clichy criteria recommend transplantation for patients in stage III or stage IV coma on the basis of age and factor V levels. The transplantation threshold is 20% factor V activity for patients <30 years or 30% factor V activity for patients >30 years.26 There is no distinction made for the etiology of ALF, which is felt to be a weakness of these criteria. The King’s College Hospital (see Table 45-5) accounted for better spontaneous outcomes of patients who had ALF on the basis of acetaminophen toxicity and divided their criteria accordingly. While the positive predictive value of King’s College Hospital criteria has been shown to be clinically acceptable in ALF patients, the negative predictive value drops below 50% in nonacetaminophen patients.27 Thus, patients who fail to fulfill these criteria include a number of patients who will die without being properly considered for transplantation. Various modifications to the King’s College Hospital criteria to improve performance and other prognostic scoring models for specific etiologies have been proposed. These include the addition of blood arterial lactate, serum phosphate, and levels of Gc-globulin, a marker that could improve quantitation of hepatic necrosis.28
Coagulopathy is also a necessary finding for the diagnosis of ALF; however, clinically significant spontaneous bleeding is uncommon. Correction of thrombocytopenia to ≥50,000/mm3 and INR to ≤1.5 is suggested for the bleeding patient or the patient about to undergo an invasive procedure.14,21 Specific treatment thresholds for the nonbleeding patient are difficult to define, but it is suggested that prophylactic therapy not be undertaken except for severe abnormalities, for example, platelet count ≤10,000/mm3, INR >7, and fibrinogen <100 mg/dL.14 Occasionally, the use of rFVIIa or prothrombin complex concentrate is used to correct a resistant INR abnormality or to avoid fluid overload. It should be kept in mind that these agents carry a thrombotic risk and are contraindicated when the etiology of ALF is associated with hypercoagulability, such as pregnancy or Budd–Chiari syndrome.
Hypotension in ALF may be the result of several days of gastrointestinal losses, poor intake, or myocardial dysfunction, but likely includes a component of decreased arterial tone as liver necrosis progresses. The hypotensive ALF patient should undergo volume status and cardiac function assessment prior to consideration of inotropes or vasopressors. Vasopressors may be used either to treat systemic hypotension or to maintain an adequate CPP. On the basis of recommendations for septic patients, either norepinephrine or dopamine may be used. The use of arginine vasopressin (AVP) or its analogs cannot be recommended as there is evidence that their use is associated with increases in ICP.29
Acute Hepatitis
The most common causes of acute viral hepatitis are, collectively, the five identified viral hepatitidies: A (HAV), B (HBV), C (HCV), D (HDV or delta-virus), and E (HEV). HAV and HBV have been well characterized and vaccines have been developed to prevent their transmission. As a result of widespread vaccination, the incidence of new cases of HAV and HBV has decreased steadily worldwide. Unfortunately, the same cannot be said for HCV, for which there is no currently available vaccine. The number of reported new cases of HCV is decreasing but this is likely the result of better screening of transfused blood products and the adoption of universal precautions. HDV is a single-strand RNA genome that requires the helper function of HBV for virion assembly and so must occur either as a coinfection or a superinfection with HBV. HEV is a small RNA virus that has been
responsible for several epidemics of hepatitis, primarily in underdeveloped countries with poor sanitation.
responsible for several epidemics of hepatitis, primarily in underdeveloped countries with poor sanitation.
The diagnosis of acute hepatitis is made on the basis of classic signs and symptoms, together with laboratory studies to assess liver damage and serologic assays. Symptoms can be nonspecific, such as fatigue, poor appetite, nausea, vomiting, and abdominal pain, and many infections are subclinical. Signs may include jaundice, or a serum-sickness–type presentation with fever, arthralgia or arthritis, and rash that results from circulating hepatitis antigen–antibody complexes. Incubation periods can be several weeks to even months and patients may undergo surgery without awareness of illness. For this reason viral hepatitis should be part of the differential diagnosis when there is any evidence of postoperative liver injury.
HAV is a picornavirus that is spread primarily by the fecal–oral route or via contaminated food or water. It is highly contagious and can be spread by close personal and household contact. HAV has a mean incubation period of 4 weeks and the virus may be shed 1 to 2 weeks before and for at least 1 week after the onset of illness. HAV has a wide range of manifestation from asymptomatic disease (particularly in children) to ALF. ALF is rare (<1%) and is more likely than other causes to result in spontaneous recovery (69%) in patients without underlying liver disease.30 There is no chronic disease state associated with HAV.
HBV is a DNA virus that is spread via parenteral, cutaneous, or mucosal exposure to infected blood or body fluids. It is an extremely hardy virus and can remain viable for days outside the body.31 The United States has a low rate of infection and the most commonly identified risk factors are parenteral drug use and sexual contact. In highly endemic populations such as in sub-Saharan Africa, Southeast Asia, and China, infection occurs primarily during the perinatal period. Since the progression to chronic disease is approximately 90% in this age group, perpetuation of endemicity is assured. The mean incubation period is 12 weeks, but can be as long as 20 weeks. HBV surface antigen (HBsAg) is the hallmark of active HBV infection and usually appears during the incubation period, 1 to 10 weeks after exposure. ALF caused by acute HBV infection occurs in less than 1% of cases, but has only a 20% rate of spontaneous recovery. Maintenance of seropositivity for HBsAg for >6 months after recovery suggests chronic infection. Progression to chronic disease occurs in 2% to 5% of infected adults. Treatment of chronic HBV is aimed at suppressing viral replication and preventing progression of liver disease. Parameters followed are serum ALT, HBV DNA levels, positive or negative status of HBeAg (a marker of viral replication), and liver histology. There are currently six therapeutic agents approved for the treatment of chronic HBV: Adefovir dipivoxil, interferon α-2b, pegylated interferon α-2a, lamivudine, entecavir, and telbivudine.
HDV infection occurs in conjunction with HBV infection and is estimated to be present in 5% of patients with chronic HBV. Two types of HDV infection are described: Coinfection with acute HBV and superinfection on top of chronic HBV. Both types cause severe infection and may cause ALF. Suspicion should be raised for HDV infection in the patient who seems to have a fulminant course of acute HBV infection or who has had stable chronic HBV disease and acutely decompensates. The course of the patient with coinfection may be biphasic, with one peak in aminotransferases reflecting peak HBV replication and another one a few weeks later reflecting peak HDV replication. HDV superinfection can be differentiated from a flare of chronic HBV by testing for HDV RNA or IgM anti-HDV.
HCV, once referred to as non-A, non-B hepatitis until its identification in 1989, is transmitted primarily parenterally. Since the identification of HCV, the ability to serologically screen blood products for its presence has all but eliminated it as a source of posttransfusion hepatitis. Causes of transmission are often not identifiable, but the most commonly known risk factor is parenteral drug use. Incubation period averages 7 weeks, jaundice is infrequent, and there is a high rate of subclinical infection. Interestingly, patients who develop jaundice are more likely to recover completely. HCV has a high rate of progression to chronic disease (50% to 85%) and a risk of developing cirrhosis ranging from 5% to 25% over 25 to 30 years.32 Contributing factors to the development of cirrhosis include older age, alcohol consumption, an immunosuppressed state, and obesity. HCV is currently the leading cause for liver transplantation in the United States, which is performed for cirrhosis and/or associated HCC. The optimal therapy of chronic HCV infection is the combination of pegylated interferon-α and ribavirin, with the goal of sustained viral response (SVR), defined as the absence of HCV RNA from serum by a sensitive PCR assay 24 weeks following discontinuation of therapy. Unfortunately therapy is often compromised by intolerance of drug-related constitutional or neuropsychiatric symptoms or by lab abnormalities such as neutropenia or hemolytic anemia.
Alcoholic Hepatitis
Alcoholic hepatitis is the syndrome marked by the development of jaundice and liver dysfunction in the setting of heavy alcohol use. The typical patient is a middle-aged man who has a history of excessive drinking and who has become acutely ill with the dramatic onset of jaundice. Other signs include fever, ascites, and abdominal tenderness, frequently the result of an enlarged tender liver. Encephalopathy may be present in severe alcoholic hepatitis and, if so, portends a poorer prognosis. Laboratory studies show moderate serum aminotransferase elevations (<300 IU/mL), with AST being elevated more than ALT. The AST:ALT ratio is >2 in about 70% of patients with alcoholic hepatitis.33 The white blood count, serum bilirubin, and INR are also elevated. Elevations of serum creatinine are particularly ominous as they may indicate impeding hepatorenal syndrome (HRS). Acute kidney injury, defined as at least a 0.3 mg/dL or 50% increase in serum creatinine over baseline, is associated with a significantly increased 90-day mortality (63% vs. 7%) in patients with alcoholic hepatitis.34
The differential diagnosis of the patient with the clinical and laboratory findings described includes nonalcoholic steatohepatitis (NASH), viral hepatitis, DILI, fulminant autoimmune liver disease or Wilson’s disease, severe ascending cholangitis, and hepatic abscess. A history of excessive alcohol use is supportive of the diagnosis of alcoholic hepatitis, but up to 20% of these patients may have a coexisting cause of liver disease.35 While liver biopsy is not required to make the diagnosis of alcoholic hepatitis, it is important to investigate other potential causes of acute liver disease. Viral hepatitis serologies should be sent, as well as autoimmune markers and ceruloplasmin. Blood, urine, and ascitic fluid cultures should be obtained. Imaging should be done to rule out space-occupying hepatic lesions or biliary processes.
The key component of therapy for alcoholic hepatitis is abstinence. For those patients with severe alcoholic hepatitis, medical therapy should also be considered. This consists of nutritional therapy that takes into account not only protein-calorie nutrition, but vitamin and mineral deficiencies as well. The administration of a 28-day course of prednisolone is also recommended for those patients with advanced disease. In patients for whom steroids are contraindicated, consideration should be given to pentoxifylline, a phosphodiesterase inhibitor with anticytokine activity. In a randomized placebo-controlled trial the administration of
pentoxifylline was associated with 40% less mortality than placebo in patients with severe alcoholic hepatitis.36
pentoxifylline was associated with 40% less mortality than placebo in patients with severe alcoholic hepatitis.36
Drug-induced Liver Injury
Often considered as an afterthought when a patient presents with new abnormalities in liver-related laboratory studies, DILI is a significant cause of morbidity and mortality. Although the process of diagnosing DILI is not well defined and it is largely a diagnosis of exclusion, DILI should always be considered when formulating the differential diagnosis of patients presenting with liver abnormalities. Moreover DILI is a serious problem for the pharmaceutical industry, as it is the most common reason for regulatory actions such as failure of approval, removal from market, or restrictions on indications for use. Nonacetaminophen drug-induced idiosyncratic liver injury accounts for 11% to 13% of cases of ALF and, with a 20% rate of survival with supportive care, has a poorer than average rate of spontaneous recovery.13,37
A recent report from the international DILI Expert Working Group has defined laboratory criteria for diagnosing DILI (Table 45-6). DILI can further be characterized as hepatocellular, cholestatic, or mixed, on the basis of the relative abnormalities of laboratory values. This is done by calculating the R value, such that R = (ALT/ULN)/(ALP/ULN), where ALT is the alanine aminotransferase, ALP is the alkaline phosphatase, and ULN is the upper limit of normal value. The higher the R value, the more abnormal the ALT in comparison to the ALP. Thus R values ≥5 are used to define a hepatocellular pattern of damage. R values ≤2 define a cholestatic pattern, and R values between 2 and 5 define a mixed pattern. A prognostic rule of thumb is eponymously named “Hy’s law” after Hyman J. Zimmerman, a leader in DILI research. It was his observation that jaundice (defined as bilirubin >2 ULN) in patients with hepatocellular DILI carried a poor prognosis, with a mortality of >10%. This observation has been confirmed and recognized for many years by the FDA as a tool for identifying which drugs may be expected to cause significant hepatotoxicity.38
Table 45-6. Clinical Chemistry Criteria for Drug-Induced Liver Injury (DILI) | |||
|---|---|---|---|
|
The liver is commonly involved in drug toxicity because of its central role in drug metabolism. Drugs may either be directly hepatotoxic or may propagate toxic metabolites, most often as products of phase I drug metabolism and the cytochromes P450.39 Cell injury follows via cell stress, mitochondrial injury, or immune-mediated injury. Cell stress may result from glutathione depletion or the binding of reactive metabolites to intracellular enzymes, proteins, or lipids. Mitochondrial injury may result from the uncoupling of mitochondrial respiration with the depletion of ATP and accumulation of reactive oxygen species (ROS). Immune-mediated injury may result from the binding of reactive metabolites to cell structures, creating antigenic entities that can invoke the formation of antibodies against the cell structures themselves.
In anesthesiology perhaps the best known potentially hepatotoxic drug is halothane. Halothane was introduced to patient care in 1956 and, because of its clinical advantages of lack of flammability, potency, and patient tolerance of administration, rapidly enjoyed widespread use. However, reports of postoperative liver injury began to appear shortly thereafter, and by 1963, over 300 cases of “halothane hepatitis” had been reported.40 The National Academy of Sciences produced a retrospective epidemiologic study on the use of halothane from these reports. The National Halothane Study reviewed cases of fatal hepatic necrosis occurring within 6 weeks of the administration of a general anesthetic, from among 34 centers in the United States. Of the 856,000 anesthetics reviewed, about 255,000 involved halothane, and 82 cases of fatal hepatic necrosis were identified. Sixty-three of these cases could be ascribed to an identifiable clinical factor, leaving 19 with otherwise unexplained hepatic necrosis. Fourteen of the nineteen had received a halothane anesthetic, but did not have consistent histologic findings. The lesions seen were similar to those seen with fatal viral hepatitis or some forms of drug-induced hepatitis.41 Uncertainty over the direct association between halothane and the cases of fatal hepatic necrosis, together with the calculated incidence of 1 in 35,000 anesthetics even if such association did exist, led to the conclusion that halothane overall had a good safety record. The possible association with repeated exposure to halothane did not go unrecognized, and there was an editorial recommendation that halothane be avoided in patients with a history of unexplained fever and jaundice following a general anesthetic.42
The typical presentation of halothane hepatitis is one of nonspecific symptoms of anorexia, malaise, and fever in conjunction with a recent exposure. More specific signs include a rash and jaundice that appears 4 to 7 days after anesthetic exposure, but may be delayed for several weeks. Laboratory studies may reveal abnormalities typical of hepatocellular damage in DILI: Significant elevations in serum transaminases (500 to 2,000 IU/L), but AP elevations that are generally no more than twice the upper limit of normal.40,43 Risk factors include female gender, obesity, age, and most important, a history of prior exposure.
It is generally agreed that halothane hepatitis is composed of two different manifestations. A relatively mild, self-limited form is characterized by elevations in liver-related lab studies without evidence of liver failure. This may occur in up to 20% of patients after halothane exposure.44 A proposed mechanism for this hepatocellular damage is the combination of halothane degradation products and hypoxia caused by imbalance in the hepatic oxygen supply–demand relationship.45 There is strong evidence that the severe, fulminant form of halothane hepatitis is an immune-mediated process. The association with repeated halothane exposure and the appearance of rash and eosinophilia support this hypothesis. Furthermore, circulating IgG antibodies against liver proteins, modified by the reactive trifluoroacetyl (TFA) metabolite
of halothane, have been identified in the sera of patients with clinical halothane hepatitis.46 Although other halogenated inhalational anesthetics that produce TFA metabolites such as enflurane, isoflurane, and desflurane have been associated with acute hepatic failure, the incidences of hepatitis attributed to them have been very small. Since halothane is by far the most extensively metabolized of these agents (20% halothane metabolized vs. 2% enflurane, 0.2% isoflurane, and 0.01% desflurane) the production of TFA metabolites would seem to correlate with the incidence of associated hepatitis. Indeed, an animal study examining the extent of hepatic tissue trifluoroacylation after exposure to halogenated anesthetics showed that halothane produced significantly more tissue acylation than enflurane, isoflurane, or desflurane.47
of halothane, have been identified in the sera of patients with clinical halothane hepatitis.46 Although other halogenated inhalational anesthetics that produce TFA metabolites such as enflurane, isoflurane, and desflurane have been associated with acute hepatic failure, the incidences of hepatitis attributed to them have been very small. Since halothane is by far the most extensively metabolized of these agents (20% halothane metabolized vs. 2% enflurane, 0.2% isoflurane, and 0.01% desflurane) the production of TFA metabolites would seem to correlate with the incidence of associated hepatitis. Indeed, an animal study examining the extent of hepatic tissue trifluoroacylation after exposure to halogenated anesthetics showed that halothane produced significantly more tissue acylation than enflurane, isoflurane, or desflurane.47
Pregnancy-related Liver Diseases
Abnormalities in liver studies occur in 3% to 5% of pregnancies. Although many causes reflect underlying hepatic or biliary disease, the most common causes are one of the five acute, pregnancy-related conditions: Hyperemesis gravidarum; intrahepatic cholestasis of pregnancy; preeclampsia; preeclampsia complicated by hemolysis, low platelet count, and elevated liver enzymes (HELLP syndrome); and acute fatty liver of pregnancy (AFLP; Table 45-7). Hyperemesis gravidarum is a feature of the first trimester of pregnancy and is characterized by vomiting of sufficient severity to warrant intravenous (IV) hydration. Risk factors include hyperthyroidism, molar pregnancy, and multiple pregnancies.48 Liver enzymes may be elevated in 50% of patients, with up to 20-fold elevation, but little if any elevation of bilirubin.49,50 It is important to distinguish hyperemesis from acute viral hepatitis or from drug toxicity with appropriate labs and a careful medication history. Therapy is primarily supportive and the condition usually resolves by the second trimester.
Intrahepatic cholestasis of pregnancy usually presents in the second to third trimester of pregnancy. The proposed etiology is interference with bile acid transport across the canalicular membrane, resulting in elevated serum bile acid elevation and pruritus. In addition to modest increases in bilirubin (usually <5 mg/dL) aminotransferases may also be elevated up to 20-fold and serum bile acids may be elevated up to 100-fold.49 As with hyperemesis gravidarum, treatment is primarily supportive, aimed at relieving pruritus. Unlike hyperemesis, intrahepatic cholestasis of pregnancy may be associated with chronic placental insufficiency, premature labor, and sudden fetal death. Therefore, pregnancies complicated by intrahepatic cholestasis of pregnancy are considered fetal high-risk pregnancies.
The three remaining uniquely pregnancy-related conditions all present in the third trimester. Preeclampsia is diagnosed by the triad of hypertension, edema, and proteinuria. Elevation of aminotransferases is indicative of severe preeclampsia. The appearance of microangiopathic hemolytic anemia (MAHA), elevated liver enzymes, and low platelet count in the preeclamptic patient comprises the HELLP syndrome and occurs in 20% of severely preeclamptic patients. MAHA is the result of vascular endothelial injury with subsequent fibrin deposition and platelet consumption. This also leads to areas of hepatic infarction and subsequent hemorrhage, which may coalesce into large hematomas and lead
to capsular rupture and intraperitoneal bleeding. The clinical presentation is not dissimilar to severe preeclampsia, with abdominal pain, nausea, headache, hypertension, and edema. Laboratory studies show elevated aminotransferases, up to 10- to 20-fold, and modest increases in bilirubin. A peripheral smear will show the characteristic schistocytes and burr cells of MAHA. Platelet count may be used to distinguish between mild, moderate, and severe HELLP, with platelet counts of 100,000 to 150,000/mm3, 50,000 to 100,000/mm3, and <50,000/mm3, respectively.
to capsular rupture and intraperitoneal bleeding. The clinical presentation is not dissimilar to severe preeclampsia, with abdominal pain, nausea, headache, hypertension, and edema. Laboratory studies show elevated aminotransferases, up to 10- to 20-fold, and modest increases in bilirubin. A peripheral smear will show the characteristic schistocytes and burr cells of MAHA. Platelet count may be used to distinguish between mild, moderate, and severe HELLP, with platelet counts of 100,000 to 150,000/mm3, 50,000 to 100,000/mm3, and <50,000/mm3, respectively.
Table 45-7. Distinguishing Features of Intrahepatic Cholestasis of Pregnancy (ICP), the Hellp Syndrome and Fatty Liver of Pregnancy (AFLP) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Serious maternal complications include DIC, placental abruption, eclampsia, pulmonary edema, hepatic hematoma, and hepatic capsular rupture.51 Abdominal CT imaging is the preferred study to detect major hepatic complications of infarct, hematoma, or rupture. Contained hepatic hemorrhage can be managed conservatively with correction of volume deficit and coagulopathy. Manipulation of the abdomen or abdominal straining should be avoided. Capsular rupture or rapid extension of a hematoma is life threatening and demands more aggressive treatment for control of bleeding, usually emergency laparotomy. Rarely, there may be an indication for transplantation for the patient in whom bleeding cannot be controlled. Delivery is definitive therapy for HELLP syndrome, although up to 25% of patients may not present until the postpartum period. Therapy remains the same regardless of timing of presentation and most patients will rapidly resolve abnormalities after delivery.
AFLP is the result of rapid microvesicular fatty infiltration of the liver resulting in acute portal hypertension and encephalopathy. Although the exact mechanism of AFLP is unknown, there is an association between it and abnormalities in the enzymes involved in β-oxidation of fatty acids. Symptoms of abdominal pain, nausea, headache, and vomiting, together with laboratory findings of elevated aminotransferases, bilirubin, and thrombocytopenia, are similar to severe preeclampsia and HELLP syndrome. However, the AFLP patient may additionally have laboratory and clinical findings more unique to liver failure, such as hypoglycemia, elevated ammonia, asterixis, and encephalopathy. Although the definitive diagnosis is made histologically, there is usually reluctance to perform liver biopsy. Noninvasive studies such as ultrasound or abdominal CT are an option and may show increased echogenicity or decreased hepatic density consistent with fatty liver. However, they cannot be relied upon as they may be unremarkable in half of the patients with AFLP.52
Arrangements for rapid delivery should follow diagnosis of AFLP, as recovery can only follow delivery. Recovery may be prolonged in patients who are severely ill upon presentation, and there is a role for transplantation in the patient who continues to deteriorate into ALF after delivery.
Cirrhosis and Portal Hypertension
Hemostasis
Hemostasis is a dynamic process that is the product of interaction between coagulation, platelets, and fibrinolysis, resulting in the formation and revision of clot. Liver disease affects all three of these components, both quantitatively and qualitatively.
The liver is the site of synthesis for all procoagulant and anticoagulant factors, with the exception of tissue thromboplastin (III), calcium (IV), and von Willebrand factor (VIII). It is also the site for clearance of activated factors.
Cirrhotic patients are customarily considered to have a bleeding diathesis on the basis of abnormalities in conventional tests of coagulation such as PT and partial thromboplastin time (PTT). However, such tests reflect the activity of only a portion of the procoagulant factors and do not consider the concomitant decrease in anticoagulant factors, which are not customarily measured. It is the balance of procoagulant and anticoagulant forces, not the isolated measurement of either portion of the coagulation system that indicates the effective generation of thrombin. Not surprisingly, PT and PTT abnormalities correlate poorly with bleeding complications following invasive procedures, such as liver biopsy.55,56,57 In fact there is evidence that, should one account for differences in the anticoagulant levels between normal and cirrhotic patients by adding thrombomodulin (an activator of the anticoagulant protein C) to the PT assay, normal and cirrhotic patients generate the same amounts of thrombin.58 Thus, one may conclude that the decreased levels of protein C in cirrhotic patients balance the decreased levels of procoagulants, leaving thrombin generation in vivo unaltered.
Even more counterintuitive is the increasing evidence that cirrhotic patients not only have normal thrombin generation, but may actually have a procoagulant imbalance on the basis of reduced levels of anticoagulants protein C and antithrombin III, together with an increase in FVIII.59,60 Clinically, this is supported by studies reporting not only the lack of protection of liver disease against the formation of venous thromboembolism (VTE)61,62 but an increased risk of VTE formation associated with the presence of cirrhotic and noncirrhotic liver disease.63
The cholestatic diseases (e.g., PBC, sclerosing cholangitis) may eventually progress to cirrhosis, but until that happens the coagulopathy of these diseases has a different nature from that of hepatocellular dysfunction. The coagulopathy of biliary disease is characterized by functional deficiencies in the vitamin K–dependent procoagulants II, VII, IX, and X and anticoagulants protein C and protein S. Vitamin K is a fat-soluble cofactor necessary for the final step in the production of these factors: Carboxylation of the precursor produced by the liver. Bile salts are necessary for absorption of vitamin K, and impaired bile secretion in cholestasis results in vitamin K deficiency. Parenteral vitamin K can correct this deficiency and return coagulation to normal as long as the liver is still capable of manufacturing adequate amounts of factor precursors. It cannot, however, correct the coagulopathy of hepatocellular dysfunction.
Dysfibrinogenemia has been described in acute, chronic, and neoplastic liver disease and is the most common qualitative defect of coagulation factors, occurring in 70% to 80% of cirrhotics.64 Its presence does not appear to be related to the severity of hepatic dysfunction, but instead to be associated with hepatic tissue regeneration. Excess sialic acid residues on the fibrinogen interfere with the enzymatic activity of thrombin and cause abnormal
polymerization of fibrin monomers. Thus, although serum fibrinogen levels may be adequate, function is not accurately reflected.
polymerization of fibrin monomers. Thus, although serum fibrinogen levels may be adequate, function is not accurately reflected.
Platelets provide primary hemostasis by interaction with the vessel wall at the site of injury and forming a physical plug. Thrombocytopenia is a well-known feature of cirrhosis. Estimates of incidence range from 30% to 64% of chronic cirrhotics, but platelet counts below 30,000/mm3 are rare.65 Because the liver is the primary site of thrombopoietin production, decreased levels of thrombopoietin contribute. Other factors include immunologic mechanisms, direct bone-marrow suppression, and consumptive processes such as DIC. However, the primary cause is splenic sequestration in the setting of portal hypertension. Up to 90% of the platelet population may be sequestered in the spleen. Elevated levels of von Willebrand factor are felt to compensate for decreased platelets counts, augmenting the platelet–endothelial cell interaction on vessel walls.
A second function of platelets is to promote thrombin generation. Activated platelets provide negatively charged phospholipids on their surfaces, which act as receptors for the assembly of coagulation factors and thus promote coagulation. A series of assays measuring thrombin generation concluded that platelet counts below a threshold of 100,000/mm3 negatively correlated with thrombin production. It was further estimated that the minimum platelet count necessary to support near-normal thrombin generation was 56,000/mm3.58 This information provides further support to the use of platelet transfusion in the bleeding patient with platelet counts in and below that range. Platelet transfusions are not indicated in the absence of bleeding.
The fibrinolytic system limits and revises clot formation. The initial step is activation of plasmin from plasminogen by enzymes such as tissue plasminogen activator (tPA). Plasmin consumes fibrin, producing fibrin degradation products such as D-dimer. The fibrinolytic system in cirrhotic patients has many abnormalities which may account for accelerated fibrinolysis, which has a reported incidence of 30% to 46% in patients with end-stage liver disease.66,67 The liver is the site of tPA clearance, and elevated tPA levels have been noted in patients with cirrhosis.68 Furthermore, the liver is the site of synthesis for plasmin inhibitors, such as plasmin activator inhibitor-1 (PAI-1) and thrombin-activatable fibrinolysis inhibitor (TAFI). However, as with the process of coagulation, what matters is the balance of these factors that promote and inhibit fibrinolysis and where their net forces lie. Commonly used studies for assessing the presence and severity of accelerated fibrinolysis include the euglobulin clot lysis time (ECLT) and thromboelastography (TEG). A clot lysis index in TEG has been defined as the ratio of the clot amplitude at 60 minutes post achievement of maximum amplitude (A60) to the clot maximum amplitude (MA). A ratio of <0.85 indicates the presence of accelerated fibrinolysis and suggests the need for an antifibrinolytic agent such as epsilon aminocaproic acid or tranexamic acid in the presence of otherwise unexplained bleeding.
DIC is primarily a thrombotic diathesis, followed by widespread secondary fibrinolysis. As factors are consumed, DIC becomes a bleeding diathesis of factor and platelet deficiencies. Whether or not DIC is a feature of stable CLD is controversial. Because cirrhosis shares common lab abnormalities with DIC, standard labs cannot distinguish between consumption and decreased synthesis and so have little utility. More recent approaches to answer this question have utilized assays for substances that would be expected to be elevated as the result of excessive thrombin production, the sine qua non of DIC. These include the cleaved by-products of coagulation factor activation such as prothrombin fragment F1 + 2, fibrinopeptide A, and thrombin–antithrombin (TAT) complexes. Elevation of these would suggest that low levels of procoagulation factors are the result of consumption rather than underproduction.
It is generally agreed on the basis of examination of these special assays that overt DIC is probably not a feature of stable CLD.69 However, an entity called “accelerated intravascular coagulation and fibrinolysis” (AICF) has been described. This may be considered a low-grade consumptive process that occurs in <30% of cirrhotics, primarily in those with severe, decompensated disease.70 While it may not have immediate clinical consequence, patients who exhibit this phenomenon are considered at increased risk to progress to DIC in the presence of a known stimulus, such as sepsis or spontaneous bacterial peritonitis (SBP).
Cardiac Manifestations
At the heart of these circulatory changes is portal hypertension. Portal hypertension causes local production of vasodilators such as natriuretic peptides, vasoactive intestinal peptide, endotoxin, glucagon, and especially nitric oxide.71 Elevated production of nitric oxide has been observed to precede the formation of the hyperdynamic circulation in cirrhosis, and inhibition of nitric oxide formation has been shown to increase arterial pressure in cirrhotic patients. Furthermore, there is reduced circulatory responsivity to sympathetic stimulation primarily due to overproduction of vasodilators.72
In addition to hyperdynamic circulation the cirrhotic patient may have a combination of other cardiac functional abnormalities that are not immediately apparent in the baseline state. These abnormalities comprise four key components of a condition termed “cirrhotic cardiomyopathy.” They include (1) the aforementioned increase in cardiac output and decrease in peripheral vascular resistance, (2) systolic and diastolic dysfunction, (3) cardiac resistance to β-adrenergic stimulation, and (4) electrophysiologic abnormalities.
Historically, cirrhosis has not been associated with cardiomyopathy since the hyperdynamic circulation was presumed to reflect cardiac vigor and the few patients who had overt dilated cardiomyopathy were thought to be manifesting alcoholic cardiomyopathy. However, elevated cardiac output is only a consequence of the profound decrease in afterload resulting from the dilated peripheral circulation. Systolic incompetence is revealed by physiologic or pharmacologic stress and is manifested by an inability to increase cardiac output in response to exercise and an inability to increase ejection fraction despite an increase in end-diastolic volume. Furthermore, the severity of cardiac dysfunction seems to be directly correlated with the severity of liver disease.73
Diastolic dysfunction has been described in cirrhotic patients as well, on the basis of diagnostic echocardiographic findings of abnormalities in transmitral flow during diastole. This consists of decrement or reversal of the E/A wave ratio and prolongation of E wave deceleration time, reflecting ventricular resistance to diastolic filling. Also supportive of the presence of diastolic dysfunction is the finding of septal and left ventricular hypertrophy on echo examination. Diastolic dysfunction renders cirrhotic patients very sensitive to changes in cardiac filling making them vulnerable to both heart failure and prerenal insufficiency.
Autonomic dysfunction is another characteristic of the altered cirrhotic cardiovascular system. Chronotropic and hemodynamic incompetence in response to various challenges such as sustained handgrip, ice water hand submersion, Valsalva maneuver, and tilt table testing has demonstrated autonomic neuropathy in 43% of cirrhotic patients. Although apparently unrelated to autonomic dysfunction, prolonged Q–Tc interval is also observed in cirrhotic patients with an incidence ranging from 30% in Child’s A to 60% in Child’s C patients.74 This should be kept in mind when treating these patients with drugs known to prolong Q–T interval.
Coronary artery disease in cirrhotic patients has become an area of interest particularly as the application of liver transplantation has expanded to include older patients with comorbidities. Risk factors for coronary artery disease in cirrhotic patients are similar to those of other patient populations: Hypertension, dyslipidemia, age, gender, and obesity. However, NASH has been recognized as an increasingly important cause for transplantation and carries with it both the cardiac disease risks of its attendant maladies, obesity and diabetes, and a chronic inflammatory state. The optimal test for identifying cirrhotic patients with significant CAD is unclear. Because many of these patients cannot exercise, pharmacologic stress testing is most commonly employed. Unfortunately, studies investigating the predictive value of noninvasive functional testing, particularly dobutamine stress echocardiography, have generally shown poor sensitivity and variable quality of negative predictive value (75% to 89%).75 Thus, among liver transplantation candidates consideration should be given to proceeding with coronary angiography if the patient is judged to have a high likelihood of CAD.76 For less complex surgeries, however, this may not be warranted.
Renal Dysfunction
The hallmarks of renal dysfunction in cirrhosis are the seemingly inappropriate avid retention of sodium and free water, together with renal hypoperfusion and consequent decreased glomerular filtration. The extreme manifestation of this is the HRS, a prerenal functional abnormality that is the renal response to the circulatory abnormalities of advanced cirrhosis. Renal function is an important risk factor for mortality, a fact that is emphasized by its presence as one of only three variables used in calculating the MELD score, the primary predictor of 3-month mortality for patients on the liver transplantation waiting list.
Although the most dramatic and unique renal manifestation of CLD is the HRS, cirrhotic patients are also at high risk for more prosaic causes of renal dysfunction, such as parenchymal renal disease, sepsis, nephrotoxicity, and hypovolemia. It is important to remember that HRS is a diagnosis of exclusion and that other possible potentially treatable causes must be ruled out since therapies will differ.
Despite the fact that the cirrhotic patient’s liver disease predominates, one should be mindful of any comorbidities that exist. Glomerulonephritis and diabetic nephropathy are not infrequent findings. NAFLD, the most common nonviral cause for adult CLD, is associated with type II diabetes. Immune complex nephropathies such as IgA nephropathy and membranous proliferative glomerulonephropathy are associated with chronic hepatitis C infection.77 In addition, some underlying causes of liver failure are directly associated with renal dysfunction. These include such diseases as amyloidosis, systemic lupus erythematosus, autoimmune hepatitis, polycystic liver disease, and Alagille syndrome.
The cirrhotic circulatory system is characterized by marked sympathetic stimulation, and activation of the renin–angiotensin–aldosterone and vasopressin systems. These combine to save salt and water and reduce renal perfusion. Elevated levels of renal prostaglandins help to maintain renal perfusion. Thus, cirrhotic patients are very sensitive to the prostaglandin inhibition of nonsteroidal anti-inflammatory medications, although there is evidence for the safety of short-term administration of selective COX-2 inhibitor celecoxib.78 Aminoglycosides, angiotensin-converting enzyme inhibitors, and angiotensin receptor blockers are other drug groups associated with nephrotoxicity in cirrhotic patients. Despite the expectation that contrast administration would be nephrotoxic, there is no evidence to support that concern.79
Cirrhotic patients are at risk for hypovolemia from a number of causes, including gastrointestinal bleeding, diuretic use, and diarrhea resulting from lactulose or rifaximin administration. Unfortunately, it can be difficult to assess intravascular volume status in patients who are total-body volume overloaded, whose measured central filling pressures may reflect transmitted elevated intra-abdominal pressures because of ascites, and whose measured serum creatinine levels are poor estimates of GFR due to decreased muscle mass.80,81 Nonetheless, pursuit of a diagnosis by discontinuing diuretics and providing volume expansion with albumin can help differentiate hypovolemia from the other prerenal etiology of interest, HRS. Failure to improve creatinine in response to such measures is strongly suggestive of HRS as the underlying cause.
HRS is the renal manifestation of the systemic circulatory derangement of end-stage liver disease. It is considered a functional derangement, primarily on the basis of successful transplantation of kidneys from HRS patients.82 Although it is often invoked in the differential diagnosis of acute renal dysfunction in cirrhotic patients, it accounts for only about 23% of the cases of acute kidney injury in hospitalized cirrhotic patients.83 Nonetheless, in cirrhotic patients with ascites, the incidence of HRS is 18% at 1 year and 39% at 5 years.84
The initial event leading to HRS is portal hypertension. This leads to the local production of vasodilators, particularly nitric oxide, which in turn causes splanchnic vasodilation. Splanchnic vasodilation leads to a decrease in the effective circulating blood volume and a decrease in arterial blood pressure. These conditions combine to activate the sympathetic, renin–angiotensin–aldosterone and vasopressin systems. The net result is a severe reduction in renal perfusion and glomerular filtration with impaired free water excretion as the kidneys do their part to try to maintain circulatory homeostasis.
The generally agreed upon criteria for diagnosing HRS are those proposed by the International Ascites Club (Fig. 45-2).85 Two manifestations of HRS are recognized, called type I and type II. Although they were once considered variants of the same disorder, it has become increasingly clear that they must be treated as two different entities.
Type I HRS is characterized by rapidly progressive renal failure, typically represented by at least a doubling of serum creatinine over the course of 2 weeks in close proximity to a precipitating cause such as SBP, sepsis, gastrointestinal bleeding,
or surgical stress. Patients with type I HRS have a median survival of 2 to 4 weeks without therapy.84,86 Type I HRS is associated with failure of other organ systems, including adrenal insufficiency. Most notably; however, when type I HRS responds to medical therapy that response is usually sustained, even after withdrawal of therapy.87
or surgical stress. Patients with type I HRS have a median survival of 2 to 4 weeks without therapy.84,86 Type I HRS is associated with failure of other organ systems, including adrenal insufficiency. Most notably; however, when type I HRS responds to medical therapy that response is usually sustained, even after withdrawal of therapy.87
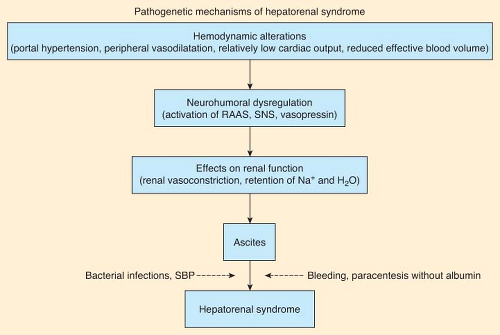 Figure 45.2. The pathogenetic mechanism of hepatorenal syndrome. Schematic view of the pathogenesis of hepatorenal syndrome in cirrhosis. Dotted arrows indicate precipitating factors that are frequently present but not necessary. RAAS, renin–angiotensin–aldosterone system; SBP, spontaneous bacterial peritonitis; SNS, sympathetic nervous system. (From: Salerno F, Gerbes A, Gines P, et al. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut. 2007;56:1312.) |
Type II HRS is more indolent and may be considered the expected consequence of continuous and progressive activity of the circulatory homeostatic triad of the sympathetic, renin–angiotensin–aldosterone and vasopressin systems in an attempt to compensate for the progressive loss of effective circulating blood volume to the increasingly dilated splanchnic vasculature. The most compelling clinical problem in these patients is refractory ascites. Patients with type II HRS have a median survival of about 6 months.88
Related posts:




Full access? Get Clinical Tree


