Chapter 90 The Innate Immune System
The immune system can be considered as a complex tool to distinguish tissues that are “infectious non-self” from “noninfectious self.”1 More recently a distinction between “dangerous” and “nondangerous” matter has been proposed as a better description for innate immunity.2
Components of the Innate Immune System
The afferent, or “sensing,” limb of the innate immune system consists of pattern recognition molecules that recognize typical molecular structures on pathogenic micro-organisms. Gram-negative lipopolysaccharide and Gram-positive lipoteichoic acid are obvious targets, but pattern recognition molecules are also directed against characteristic non-human patterns of sugars involving mannose, glucose, fructose, N-acetyl-D-glucosamine, and N-acetyl-mannosamine. Mannose-binding lectin (MBL) binds to all of these and thus recognizes a wide variety of bacteria, viruses, yeasts, fungi and protozoa, but also endogenous ligands.3
The importance of these danger signals is illustrated by the number of names competing in the literature to describe them: pathogen-associated molecular patterns, danger associated molecular patterns, and most recently alarmins.4 Whatever name used, the importance is that when such patterns encounter pattern recognition molecules of the innate immune system, they initiate and/or potentiate a cascade of events that is aimed at rapid killing of invading pathogens or reestablishing immune homeostasis.
Circulating Pattern Recognition Receptors: Complement, Lectins, and Defensins
Sugar-recognizing collectins, molecules that contain collagenous structures and C-type carbohydrate recognizing domains (CRD) include MBLs and surfactant proteins A and D (SP-A and SP-D). Mannose binding lectin is a liver derived acute phase reactant whereas SP-A and SP-D are synthesized in the lung. The main determinant of MBL levels is genotype, whereas SP-A and SP-D do increase significantly with inflammatory stress. Collectins bind to many microbes: viruses, bacteria, fungi, and protozoa and prepare organisms for phagocytosis (opsonization) and activate complement pathways. The ficolins, L-ficolin, M-ficolin, H-ficolin, are similar but they have different structures with a fibrinogen-like domain. Both MBL and the ficolins initiate the lectin pathway of complement activation via associated serine proteases.5
Alarmins, such as α and β defensins, and cathelicidins are expressed in neutrophils, intestinal Paneth cells, and epithelial cells in the respiratory tract. Defensin synthesis and release occurs constitutively but increases with cellular activation. Defensins exert a dose-dependent direct bactericidal activity and function as chemo-attractants for phagocytes. They also act generally to increase phagocytic function by increasing production of reactive oxygen species, binding to C1q to activate the classical complement pathway and inhibiting the production of immunosuppressive adrenal glucocorticoids.6
Cellular Pattern Recognition Receptors
The archetypal pathogen-associated molecular pattern is the gram-negative cell wall component lipopolysaccharide (LPS), to which humans are exquisitely sensitive. Endotoxin or LPS is recognized by the humoral factor LPS binding protein and the cell membrane receptor toll-like receptor 4 (TLR4). LPS binding protein, a liver-derived glycoprotein, binds to LPS and shuttles it to an immune cell surface (e.g., a monocyte). The monocyte receives the LPS binding protein/LPS compound on a complex consisting of TLR4, CD14, and MD2. On binding, the transmembrane receptor TLR4 signals to intracellular components, that in turn, activate nuclear factor κB (NFκB)-mediated downstream gene expression cytokine activation (Figure 90-1). Thus a gram-negative infection such as meningococcemia gives rise to a rapid nonspecific response that aims to kill the inoculum and contain the infection.
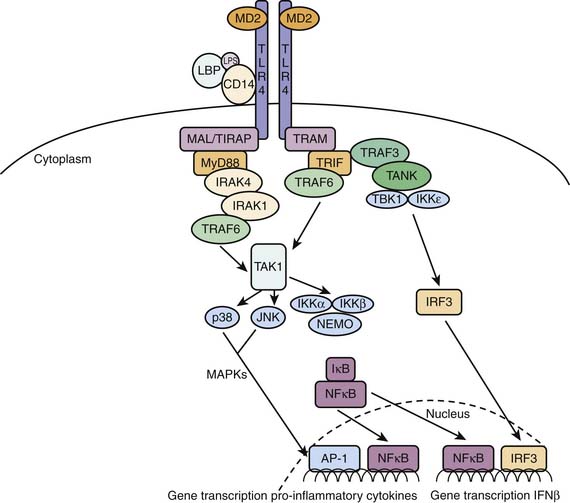
Figure 90–1 LBP shepherds LPS to the TLR4 complex. On ligation, TLR4 dimerizes and activates the two distinct MyD88- and MyD88-independent TRIF-dependent pathways. The early inflammatory phase is characterized by the MyD88-dependent pathway. This pathway activates MAPKinase and NFκB mediated pro-inflammatory gene induction. The TRIF-dependent pathway will activate late-phase NFκB and IRF3 mediated gene expression resulting in a more endotoxin tolerant response.38–40 LBP, Lipopolysaccharide binding protein; LPS, lipopolysaccharide (endotoxin); CD14, cluster of differentiation 14; MD2, myeloid differentiation protein 2; TLR4, Toll-like receptor 4; MyD88, myeloid differentiation primary-response protein 88; MAL/TIRAP, MyD88 adaptor like/Toll-interleukin 1 receptor (TIR) domain containing adaptor protein; IRAK4, interleukin 1 receptor associated kinase 4; IRAK1, Interleukin 1 receptor associated kinase 1; TAK1, transforming growth factor β-associated kinase 1; TRIF, TIR domain-containing adaptor protein inducing interferon β; TRAM, TRIF-related adaptor molecule; TRAF6, tumor necrosis factor receptor–associated factor 6; TRAF3, tumor necrosis factor receptor–associated factor 3; TANK, TRAF family member–associated NFκB activator; TBK1, TANK binding kinase 1; IKKε, inhibitor of nuclear factor κB kinase ε; IKKα, inhibitor of nuclear factor κB kinase α; IKKβ, inhibitor of nuclear factor κB kinase β; p38, p38 kinase; JNK, c-Jun N-terminal kinase; MAPKs, mitogen activated protein kinases; NEMO, nuclear factor κB essential modulator; IRF3, interferon regulatory factor 3; IκB, inhibitor κB; NFκB, nuclear factor κB; AP-1, activator protein 1.
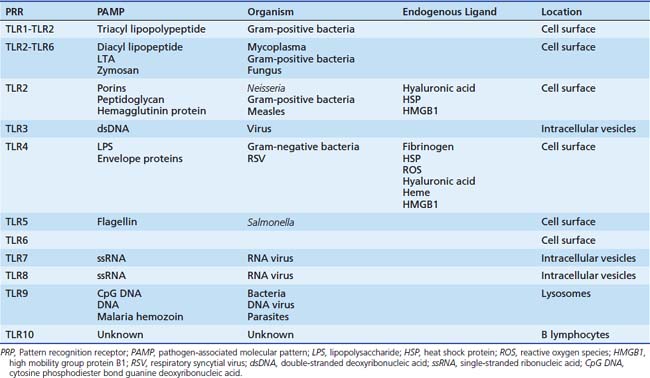
TLR4 is only one of a family of related molecules. There are at least 10 related receptors in humans (Table 90-1). These TLRs are evolutionary preserved from the worm Caenorhabditis elegans and strikingly homologous to Toll, a gene product essential to Drosophila immunity. These TLRs are transmembrane glycoproteins that are characterized by an extracellular binding domain with varying numbers of leucine-rich-repeat motifs and an intracellular cytoplasmic signaling domain homologous to that of the interleukin-1 receptor (IL-1R). This Toll/IL-1R homology (TIR) domain sits beneath the plasma membrane and interfaces primarily with the key signaling adaptor myeloid differentiation primary response gene 88 (MyD88).7
A different type of pattern recognition receptors recognize pathogens after invasion in the cytosol. RIG-I-like receptors (RLRs) respond to viral RNA. Activation induces NFκB-mediated gene transcription and production of type I interferon (INF). Of particular relevance to the intensivist are the nucleotide binding oligomerization domain and leucine rich repeat containing molecules (NLRs). The best studied of these proteins, NOD1 and NOD2, both contain N-terminal CARD domains and are specialized in detection of bacterial peptidoglycan components. Two types of activation occur: NOD1 and NOD2 ligation causes their oligomerization, that in turn induces downstream gene expression via NFκB activation. Alternatively, NLRs activate caspase-1 activating complexes, also known as inflammasomes, that in turn mature cytokines IL1-β and IL-18.8 Inflammasomes may be seen as sensors for danger. For instance, loss of cell integrity activates the inflammasome and hence the potent pro-inflammatory cytokine IL-1β. The significance of this pathway is increasingly recognized. Recent data show the inflammasome to be integral to the pathogenesis of severe Staphylococcus aureus infections.9








