1. A wide variety of disorders, with common as well as unique management principles, arise from the organs of the foregut.
2. These disorders are often associated with congenital anomalies and/or prematurity. When the associated anomalies include a severe cardiac defect, morbidity increases. Morbidity also increases in premature infants with low birth weight.
3. Tracheoesophageal fistula (TEF) and esophageal atresia (EA) are defects that may present as part of the VACTERL association. TEF/EA patients require careful attention to airway management and ventilation with the goal of prevention of aspiration.
4. Congenital pulmonary lesions (congenital lobar emphysema, congenital cystic adenomatoid malformation, and pulmonary sequestration) are associated with impaired ventilation and perfusion, leading to pulmonary and cardiovascular compromise.
5. Pyloric stenosis results in a hypochloremic metabolic alkalosis, which must be corrected preoperatively. Clinical challenges include the prevention of aspiration and electrolyte management.
6. Duodenal atresia is seen in newborn infants and is frequently associated with congenital abnormalities. Patients may be dehydrated preoperatively, and perioperative fluid and electrolyte management is of great importance.
7. Biliary atresia is the most common cause of persistent jaundice in the newborn, resulting from obstruction to bile flow and cholestasis. Prior to undergoing the hepatic portoenterostomy procedure (Kasai procedure), a careful evaluation of the patient’s coagulation status and liver function should be performed.
I. Introduction. A wide variety of congenital anomalies arise from the organs of the foregut, each associated with unique management principles. The foregut derivatives include the primordial pharynx and its derivatives, the lower respiratory system, the esophagus and stomach, the duodenum, the liver, biliary apparatus, and pancreas (1). While many children with anomalies of the foregut have isolated malformations, it is also possible for these problems to occur in association with other anomalies or as part of a syndrome that may impact anesthetic management.
II. Development of the foregut
A. Background. The primordial gut is a long tube that extends the length of the embryo and forms during the 4th week of development. The digestive tube begins cranially at the oropharyngeal membrane and ends caudally at the cloacal membrane. The endoderm of the primordial gut establishes most of the epithelium and glands of the digestive system from mouth to anus (1).
B. Divisions of primordial gut. The primordial gut is divided into three parts: the foregut, the midgut, and the hindgut. The boundaries of each of these correspond to the territories of the three arteries that supply the primordial gut (Fig. 21.1). These arteries are derived from the blood vessels that supply the yolk sac. As the yolk sac shrinks relative to the growing embryo, the left and right vitelline artery plexuses, which arise on the yolk sac, spread to form numerous arteries that supply the gut tube and anastomose with the ventral surface of the dorsal aorta. Eventually, these vessels lose their connection with the yolk sac and become the arteries that supply blood from the dorsal aorta to the digestive tract (2). Cranial to the diaphragm, approximately five aortic branches supply the thoracic part of the foregut. Caudal to the diaphragm, the remainder of the primordial gut is supplied by three arteries: the celiac artery which supplies the abdominal foregut; the superior mesenteric artery, which supplies the midgut; and the inferior mesenteric artery, which supplies the hindgut.
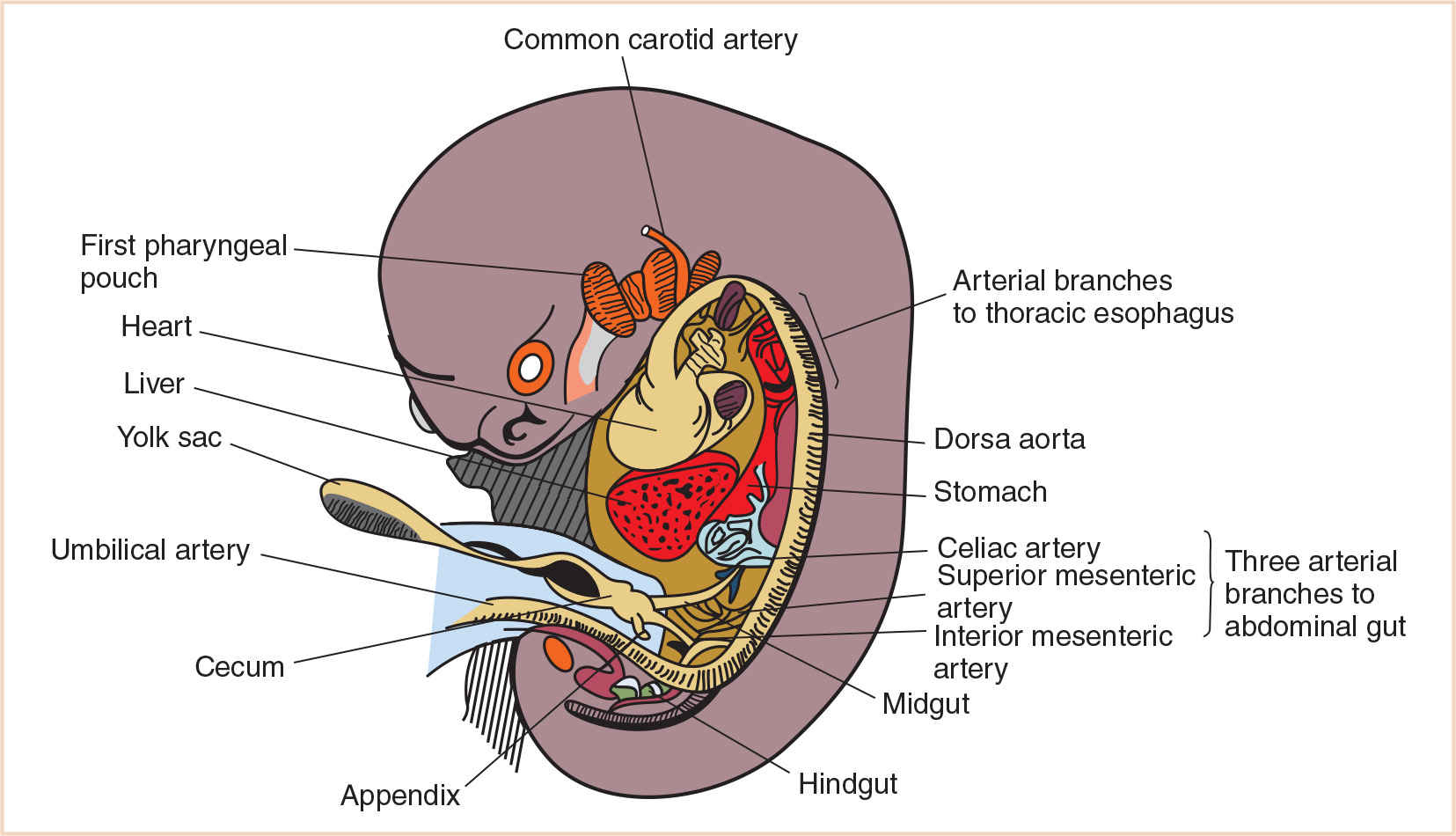
FIGURE 21.1 Ventral aortic branches supplying the primordial gut and its derivatives at 7 weeks of gestation.
C. Foregut derivatives. The foregut gives rise to (i) the pharynx and its derivatives (which include the oral cavity, pharynx, tongue, tonsils, salivary glands, and upper respiratory system); (ii) the lower respiratory system; (iii) the esophagus; (iv) the stomach; (v) the proximal half of the duodenum (which extends to the opening of the bile duct); (vi) the liver; (vii) the biliary system; and (viii) the pancreas (Fig. 21.2). Other than the pharynx, respiratory tract, and most of the esophagus, these derivatives are supplied by the celiac artery (1). Anomalies of the pharynx and upper respiratory system are discussed in detail in Chapter 16.
III. Trachea and esophagus
A. Development. The development of the upper respiratory system (the nose, nasopharynx, and oropharynx) is detailed in Chapter 16. The lower respiratory organs (the larynx, trachea, bronchi, and lungs) begin to develop as a ventral outgrowth of the foregut during the 4th week of gestation. The epithelium of the internal lining of the lower respiratory tract is entirely endodermal in origin. The connective tissue, cartilage, and smooth muscle of the respiratory tract are derived from the splanchnic mesoderm that surrounds the foregut (3).
Early in the 4th week, the respiratory primordium begins as a longitudinal groove in the median plane of the ventral wall of the primordial pharynx. This laryngotracheal groove develops just caudal to the fourth pair of pharyngeal pouches, and then deepens and protrudes to form the laryngotracheal diverticulum (4). By the end of the 4th week, this develops further into the pouch-like respiratory diverticulum (lung bud), elongating and enlarging to form a tracheal bud at its distal end.
Initially, the respiratory diverticulum communicates with the foregut; however, as the diverticulum grows and expands caudally, longitudinal tracheoesophageal folds separate it from the foregut (Fig. 21.3). These folds fuse to form the tracheoesophageal septum, which divides the foregut into a dorsal portion, the esophagus, and a ventral portion, the laryngotracheal tube (which becomes the lower respiratory tract) (Fig. 21.4). The laryngotracheal tube continues to communicate with the pharynx through the primordial laryngeal inlet (1,3,5).
The development of the larynx and its associated anomalies is described more clearly in Chapter 16. The tracheal bud continues to divide into two outpouchings, the primary bronchial buds. At this point, the trachea is recognizable as a hollow stalk. By the end of the embryonic period, the splanchnic mesoderm gives rise to the cartilaginous rings and muscles of the trachea. The endoderm has developed into the epithelium and glands. The bifurcation into the main bronchi descends from its initial cervical level to the upper thoracic level (6).
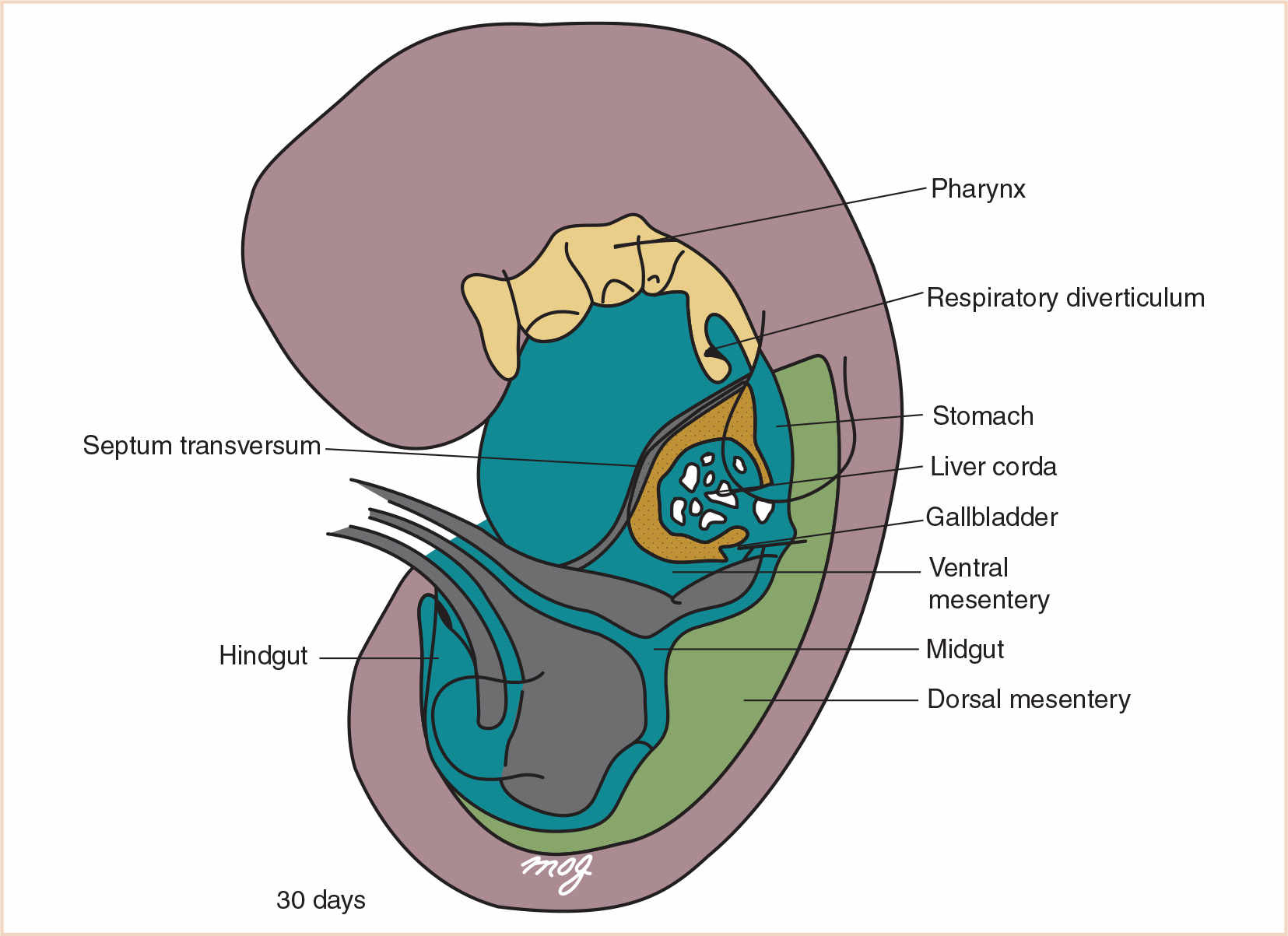
FIGURE 21.2 The primordial gut. The foregut includes the pharynx, the respiratory diverticulum, the esophagus, the stomach, the proximal half of the duodenum, the liver, the gallbladder, the pancreas, and their associated ducts.
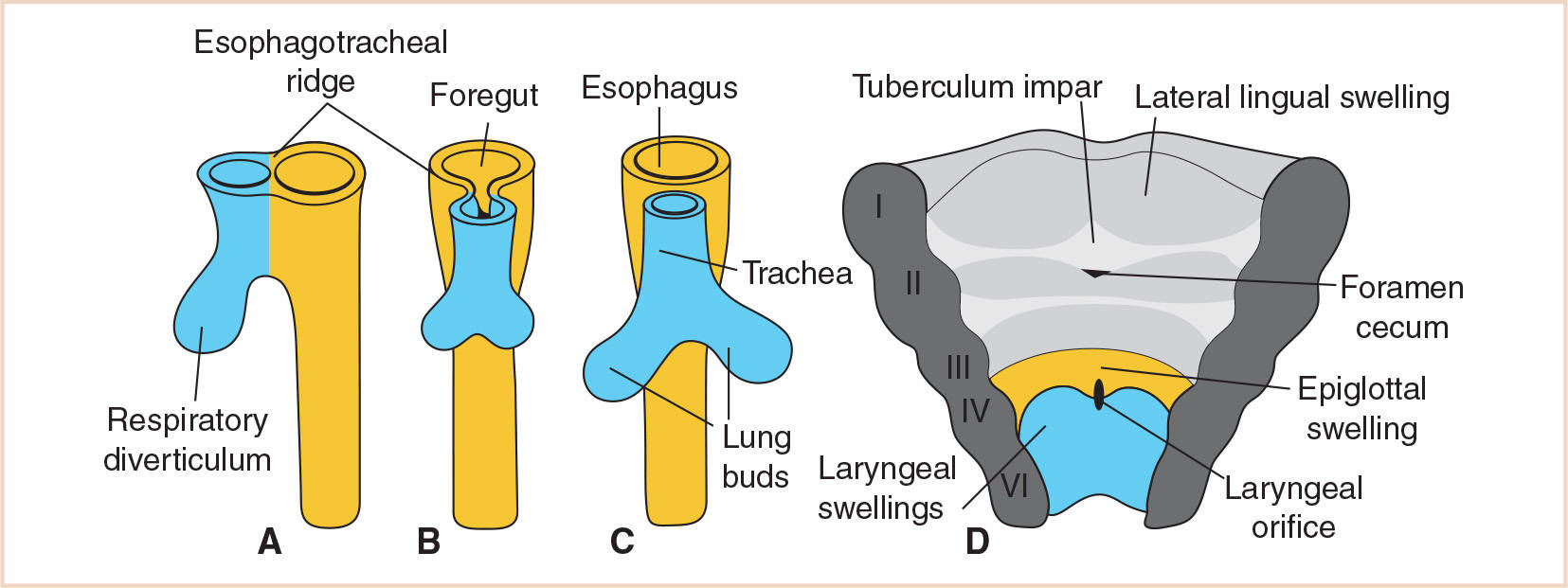
FIGURE 21.3 Stages in the development of the respiratory diverticulum. Esophagotracheal ridges fuse to form the tracheoesophageal septum and separate the foregut into the esophagus and trachea. The ventral wall of the pharynx shows the primordial laryngeal orifice.
The esophagus now lies immediately caudal to the pharynx. Initially the esophagus is very short, but with the descent of the heart and lungs it elongates rapidly. By the 7th week of development, the esophagus has reached its final relative length. The epithelium, which is derived from the endoderm, proliferates and is thought to either partly or completely obliterate the lumen of the esophagus. However, by the end of the embryonic period, recanalization occurs (1). The fetus is able to swallow amniotic fluid, which passes to the intestine where it is absorbed and transferred through the placenta to the maternal blood. If the fetus is not able to swallow at this point, polyhydramnios may develop. The superior two thirds of the esophagus develop striated muscle derived from mesenchyme in the caudal pharyngeal arches. The inferior one-third consists of smooth muscle derived from the surrounding splanchnic mesenchyme.
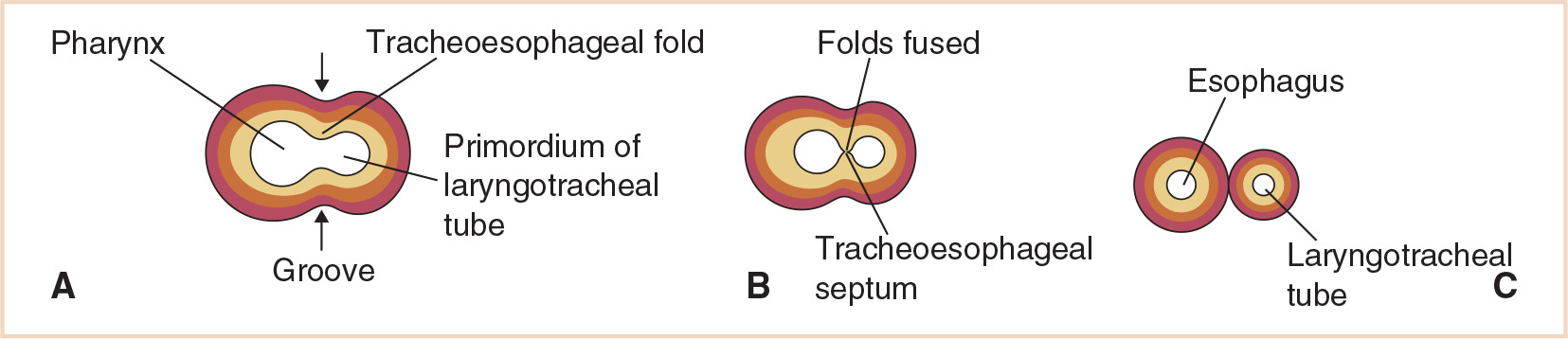
FIGURE 21.4 Development of the tracheoesophageal septum showing the eventual separation of the laryngotracheal tube and esophagus.
B. Tracheoesophageal fistula, esophageal atresia
1. Background. The incidence of tracheoesophageal fistula (TEF) is between 1:3,000 and 1:4,000 live births (7), with more than 85% associated with esophageal atresia (EA). This defect is slightly more common in males, and approximately 20% to 30% of babies with TEF are born prematurely. Many affected infants, especially those with isolated EA, have additional congenital anomalies. As many as 35% of infants with TEF have associated congenital heart disease such as: ventricular septal defect, atrial septal defect, tetralogy of Fallot, atrioventricular canal, and coarctation of the aorta. Other associated anomalies include gastrointestinal disorders, musculoskeletal anomalies, and central nervous system abnormalities (7). The survival rate of patients with TEF/EA has increased over the past few decades, with the greatest risk factors for increased mortality being birth weight <2,000 g or severe associated cardiac anomalies (8).
2. VACTERL. The likelihood of coexisting anomalies is greatest in cases of isolated EA and least with isolated TEF. The VACTERL association is the most widely recognized with TEF/EA.
a. The VACTERL association occurs when three or more of these anomalies are present.
• Vertebral anomalies: hemivertebrae, vertebral fusion, scoliosis
• Anorectal anomalies: imperforate anus, cloacal deformities
• Cardiac anomalies: includes all the defects listed earlier
• Tracheoesophageal fistula
• Esophageal atresia
• Renal anomalies: renal agenesis, renal dysplasia, horseshoe kidney, polycystic kidneys, urethral atresia, ureteral malformations, defects of external genitalia
• Limb anomalies: radial dysplasia, absent radius, syndactyly, polydactyly
CLINICAL PEARL Approximately 25% of all patients with EA have VACTERL. Therefore, if any of the anomalies mentioned above are present, diagnostic testing must be performed to evaluate for the other possible anomalies. A preoperative echocardiogram to assess for cardiac anomalies and evaluate the aortic arch, as well as a preoperative renal ultrasound can help identify the presence of these lesions, which when present may affect anesthetic management.
b. TEF/EA has a less common relationship with the CHARGE association, which consists of colobomas of the eye, heart defects, atresia of the choanae, retarded growth or central nervous system anomalies, genital hypoplasia, and ear anomalies or deafness.
3. Embryology and anatomy. Incomplete fusion of the tracheoesophageal folds results in a defect in the tracheoesophageal septum, and failure of the tracheoesophageal septum to successfully partition the foregut into the esophagus and trachea results in the development of a TEF. This septation process is thought to start caudally at the carina and extend in a cephalad direction. By 26 days of gestation, the trachea and esophagus should have separated completely up to the level of the larynx. EA is believed to result from deviation of the tracheoesophageal septum in a posterior direction. Isolated EA may result from failure of recanalization of the esophagus during the 8th week of development due to a defect in the growth and apoptosis cycle of endodermal cells (1).
While interruption of the septation process is an attractive theory, it has not been supported by findings in human embryology studies (9,10). Furthermore, rats exposed to doxorubicin (adriamycin) during early gestation have been shown to develop a variety of anomalies including TEF and EA (11,12). This adriamycin model suggests that the notochord may play an important role in the development of the trachea and esophagus, as well as an explanation for why vertebral anomalies often accompany TEF and EA. While the embryogenesis of the trachea and esophagus are understood, the molecular mechanism that causes the development of TEF and EA are not well understood (13). Hedgehog proteins are glycoproteins that are known to be involved in foregut embryogenesis. Mutant sonic hedgehog proteins have been found in patients with TEF and/or EA, and have been implicated in its pathogenesis (14).
4. Classification. The most commonly cited classification of TEF was developed by Gross, who described types A through F. Of these, type C, in which EA occurs with a distal fistula slightly above the carina, is by far the most common, occurring in approximately 90% of cases (Fig. 21.5). Isolated EA (type A) occurs in 4% to 7% of cases. The other types are less common (7).
5. Clinical features. Most cases of TEF/EA are not diagnosed prenatally, but should be suspected in cases of maternal polyhydramnios, which results from the inability of the fetus to swallow amniotic fluid. In the majority of cases, the diagnosis is made in the immediate neonatal period. The infant may be unable to swallow secretions and appear to drool excessively. Significant coughing, respiratory distress, and cyanosis may occur as a result of antegrade aspiration as the blind esophageal pouch fills (15). In patients with a distal TEF, the stomach may fill with air allowing gastric secretions to reflux back into the lungs. The diagnosis of EA is often confirmed by the inability to pass a catheter from the esophagus into the stomach. A radiograph may reveal a catheter in the blind esophageal pouch. The presence of gas in the stomach and small intestine on chest or abdominal film establishes the presence of a distal TEF.
6. Physiologic considerations
a. Airway management and ventilation is the crucial issue. Excessive positive pressure ventilation (PPV) can be quite detrimental to these patients. Ineffective ventilation can also be problematic, and may be due to placement of the endotracheal tube (ETT) in or above the fistula, obstruction of the ETT lumen with a mucous plug or clotted blood, or an excessive leak due to an inadequate seal.
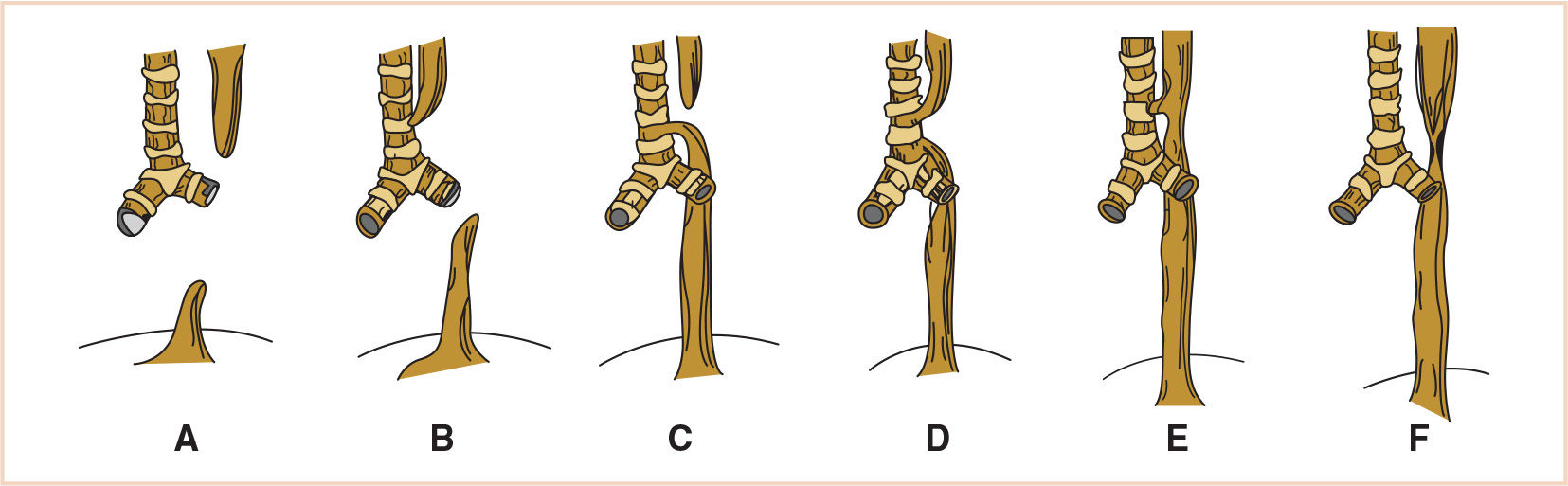
FIGURE 21.5 The Gross classification of congenital anomalies of the trachea and esophagus. A: EA without fistula. B: EA with proximal fistula. C: EA with distal fistula. D: EA with proximal and distal fistula. E: TEF with no EA. F: esophageal stenosis. EA, esophageal atresia; TEF, tracheoesophageal fistula.
b. Gastric dilation. Malposition of the ETT combined with PPV may also lead to gastric dilation and subsequent aspiration. Gastric distension may also impair ventilation.
c. Severe preexisting lung disease from either aspiration of gastric contents or respiratory distress syndrome (RDS) due to prematurity.
d. Pathophysiology of associated anomalies, particularly cardiac anomalies (16).
e. Prematurity considerations: hypoglycemia, hypocalcemia, low birth weight, hypothermia, apnea/bradycardia, and anemia (7).
7. Surgical repair: TEF
a. Primary repair without gastrostomy is typical for most healthy neonates with uncomplicated histories and proximal EA with distal fistulas. Gastrostomies as part of the primary repair or as a preliminary procedure in complicated cases are still performed, and are the initial procedures as a sequence of staged procedures in eventual repair of more complicated anomalies or sicker infants. The gastrostomy may decrease the risk of aspiration in the short term, but increase the risk of reactive airway disease in the long term by reducing lower esophageal sphincter competency and altering gastroesophageal junction geometry, thus predisposing the patient to chronic aspiration.
b. Rigid bronchoscopy immediately before surgical repair is useful to confirm the diagnosis and position of the fistula. It also allows for occlusion of the fistula with a Fogarty balloon catheter, which is placed via bronchoscopy through the fistula into the esophagus. Adequate occlusion of the fistula minimizes the likelihood of gastric distension and allows for gentle PPV intraoperatively. Once the fistula is ligated, the balloon is deflated and the Fogarty catheter is removed.
c. The surgery is typically performed in the left lateral decubitus position through a right thoracotomy. In the case of a right-sided aortic arch, a left thoracotomy would usually be performed. The approach may be extrapleural or transpleural. The fistula is divided and the tracheal side is closed transversely. Division of the fistula may dramatically improve ventilation if the fistula was not previously occluded using the Fogarty catheter.
d. An alternative to thoracotomy is the minimally invasive thoracoscopy technique. This approach provides the surgeon excellent visualization and magnification, but is technically challenging. The benefit of avoiding a posterolateral thoracotomy may lead to better cosmetic results and fewer musculoskeletal problems, but may present intraoperative ventilation challenges (which are even more problematic in the congenital cardiac population), and result in an increased operating time. The possible benefits of less postoperative pain and a shorter hospital stay have not been confirmed (17).
8. Surgical repair: EA. Surgical strategies include immediate primary repair, delayed primary repair, and esophageal replacement.
a. Immediate primary repair. Most infants with EA/TEF are candidates for immediate primary repair without gastrostomy. A single layer end-to-end primary anastomosis between the proximal esophageal pouch and the distal esophageal segment is performed once the TEF is repaired. In most patients, a gap exists between the proximal and distal esophagus requiring mobilization of both ends.
b. Delayed primary repair. This is often carried out in patients that have a long-gap EA, though the size of the gap between esophageal pouches is difficult to quantify for a variety of reasons. Surgeons may find a variety of gap lengths amenable to primary closure, and measurement of gap length is not consistently performed (methods vary from radiologic measurements to direct measurements either before or after mobilization). Therefore, there is no precise definition of “long gap” EA (8), but for the purposes of discussion, long-gap patients are those requiring staged repair. In cases of isolated EA, there is almost always a substantial gap between the esophageal ends. If primary repair is not feasible, a gastrostomy tube is placed and primary repair is delayed for up to 3 months during which time there may be growth of the upper esophageal pouch.
c. Serial dynamic lengthening. As a general rule, the patient’s own esophagus is preferable to any replacement. Preliminary traction on the atretic esophageal ends allows subsequent primary anastomosis (18,19). More specifically, the Foker technique involves the placement of traction sutures on both proximal and distal esophageal segments. Traction sutures exit the chest wall and are serially pulled in opposite directions over a period of 1 to 2 weeks, until the esophageal pouches approximate and can be anastomosed (8).
d. Other maneuvers that may facilitate anastomosis in long-gap EA include myotomy of the upper pouch, gastric pull-up, division of the lesser curvature, and greater curve elongation (20).
e. Esophageal replacement. If the previous measures fail, two options are available. First, the more conventional approach is to perform a cervical esophagostomy and esophageal replacement using colon or stomach at 12 to 18 months of age. Second and more recently, esophageal replacement with gastric interposition is performed during the initial thoracotomy (21).
9. Anesthesia conseiderations
a. Preoperative issues
(1) Minimize risk of aspiration. Patients with these lesions have typically been NPO since diagnosis. Infant should be kept in a semi-upright position, and the upper esophageal pouch should be continuously suctioned.
(2) If respiratory distress and hypoxemia exist preoperatively regardless of etiology (aspiration most likely), then urgent tracheal intubation and mechanical ventilation may be necessary. Respiratory failure may be due to pulmonary aspiration, RDS, gastric distension, or congestive heart failure. The endotracheal tube (ETT) should be positioned carefully to avoid gastric distension and aspiration, which is best accomplished if the tip of the ETT is distal to the fistula in patients with TEF.
(3) Rule out associated congenital anomalies, especially cardiac defects. An echocardiogram should be obtained along with a chest X-ray and renal ultrasonography. The presence of a right-sided aortic arch (5%) must be identified because a left thoracotomy approach will be required; the thoracotomy approach is the side opposite to the aortic arch.
(4) Preoperative laboratory tests should include glucose, hematocrit, serum electrolytes, blood urea nitrogen (BUN), and creatinine.
b. Anesthesia goals
(1) Avoidance of excessive PPV. This is especially important before the placement of the Fogarty balloon catheter or the ligation of the fistula. Stomach distension due to high airway pressures will decrease the functional residual capacity (FRC), impair ventilation, and oxygenation, and increase the chance of aspiration.
(2) Accurate positioning of the ETT. The tip should be beyond the opening of the fistula in the trachea but above the carina. This can be extremely challenging because the distal fistula (seen in type C) is often very close to the carina. The fistula may also occur distal to the carina. ETT positioning must always be rechecked following changes of patient position (i.e., supine to lateral) because of the small distances involved between the fistula, the tip of the ETT, and the carina. Tube movement distally or proximally with patient repositioning and movement of the head and neck is common.
(3) Prevention of aspiration. This is accomplished by avoidance of gastric distension during PPV.
(4) Achieve adequate perioperative pain control. This may be achieved in a variety of ways as discussed below.
c. Management of anesthesia for TEF/EA: The patient will be in the left lateral decubitus position for a right thoracotomy (left thoracotomy if right-sided aortic arch). The typical surgical time is 2 to 4 hours for primary repair.
(1) Equipment and monitoring. Equipment should include a low compression volume anesthesia breathing circuit (circle absorption system vs. Mapleson D vs. Bain circuit). Monitoring: Standard noninvasive monitoring (may include pre- and postductal pulse oximeters); arterial line for continuous blood pressure monitoring and analysis of blood gases; precordial stethoscope is useful in detecting intraoperative airway obstruction (22). Placement of two adequate peripheral intravenous (IV) lines.
(2) Induction
(a) Suction the proximal esophageal pouch before induction.
(b) A safe approach for managing the airway is an inhalation induction with or without muscle relaxation with careful, gentle PPV (no >10 to 15 cm H2O). Awake intubation is an alternate technique for securing the airway; however, oropharyngeal trauma and increases in intracranial pressure must be considered in a vigorous neonate (22). A rise in intracranial pressure may contribute to the occurrence of intraventricular hemorrhage in premature infants.
(c) Once the infant is adequately anesthetized, the surgeon is able to perform rigid bronchoscopy with a ventilating bronchoscope, following removal of the ETT. At this point, the exact location and size of the fistula can be determined and it can be occluded using a Fogarty balloon catheter. The ETT can then be replaced under direct visualization.
(d) In cases where bronchoscopy is not performed, other means are necessary to confirm the position of the ETT. After induction, an intentional right mainstem intubation may be followed by the slow withdrawal of the ETT until breath sounds are heard on the left. The use of a cuffed ETT may minimize the risk of either gastric distension or aspiration in this scenario (23). It is recommended to confirm the position of the ETT with direct fiberoptic visualization. While confirming ETT position radiographically is also an option, this method of confirmation may not ensure that the ETT is below the fistula.
(3) Maintenance of anesthesia
(a) A balanced anesthetic is recommended using either an inhalation agent/opioid technique or a combined general and epidural anesthetic consisting of an inhalation agent/thoracic epidural blockade technique. Nitrous oxide is best avoided. FIO2 should be carefully monitored to maintain a preductal SpO2 of 95% to 100%.
(b) Healthy infants may tolerate spontaneous ventilation, but most often neuromuscular blockade is necessary especially once the chest is opened and the lungs are retracted. It can be difficult to obtain adequate oxygenation, ventilation, and surgical conditions in a spontaneously breathing patient during open thoracotomy (16).
(c) Gentle PPV with minimal peak inspiratory pressure (PIP) to avoid gastric distension. Manual ventilation allows for the adjustment of ventilation during surgical retraction of the lung and also continuous direct monitoring of lung compliance.
(d) Ventilation difficulties may be encountered intraoperatively. Hypoxia/hypercarbia may be caused by retraction of the lung, kinking of the trachea or ETT from surgical manipulation and traction, occlusion of the ETT by blood clots or secretions, and malposition of the ETT. Hypercarbia may be particularly problematic in thoracoscopic repairs. These problems may be poorly tolerated by the infant with preexisting lung disease.
(e) Blood loss is usually minimal. Volume support with colloid and crystalloid is usually adequate without the need for blood products unless the infant was anemic preoperatively (which may occur in a premature infant). Maintain hematocrit >35%. In general, the judicious use of IV fluids is recommended to minimize the risk of fluid overload and pulmonary edema.
(f) Blood gas monitoring is recommended to check pH, Po2, Pco2, hematocrit, glucose, electrolytes, and possibly coagulation status.
(g) Maintain body temperature intraoperatively. The infant is prone to hypothermia from infusion of cold IV fluids, cool ambient room temperature, and high-flow anhydrous gas delivery.
(h) Ten percent dextrose-containing infusion should be continued intraoperatively because of the minimal glycogen stores in neonates and the risk of hypoglycemia.
(4) Emergence
(a) In an otherwise healthy infant requiring a straightforward ligation of a TEF, extubation in the operating room (OR) may be possible, but this is not routine. Reintubation may be necessary due to tracheomalacia or a defect in the tracheal wall at the site of the repair (21). In addition, reintubation may compromise the repair.
(b) Most infants remain intubated postoperatively and are transported to the neonatal intensive care unit (NICU).
d. Postoperative care
(1) Most infants require postoperative ventilation for a minimum of 24 to 48 hours. Those infants who have had repair of a long-gap EA usually require intubation and ventilation for 5 to 7 days (7). Any patient with preexisting lung disease, cardiac disease, or a complicated repair may require prolonged ventilatory support. On the other hand, term infants without significant comorbidities can be extubated at the end of surgery and may indeed do better without an ETT abutting the less competent tracheal cartilage. This decision must be made on the basis of the availability of excellent perioperative critical care support with the goal of avoiding reintubation under emergency conditions.
(2) Nasopharyngeal and oropharyngeal suctioning catheters should be carefully marked to avoid insertion down to the level of the anastomosis.
(3) Excessive extension of the neck should be avoided to minimize tension on the anastomosis.
(4) Pain management is most commonly accomplished with epidural analgesia or continuous narcotic infusion. Thoracic epidural analgesia may be ideal, providing a more expeditious progression to spontaneous ventilation and extubation. Recently, paravertebral catheters have also been used to manage postoperative pain after infant thoracotomies, with good results (24).
e. Regional anesthesia
(1) Epidural analgesia with placement of a thoracic epidural may be appropriate if the patient has no associated vertebral anomalies. If epidural placement is planned, chest and abdominal radiographs should be examined to rule out vertebral defects. The epidural catheter can be inserted at the thoracic or lumbar level, or may be placed transcaudally and advanced to the appropriate thoracic level. The position of the tip of the catheter can be confirmed radiographically (25).
(2) Paravertebral catheters have been increasingly used in the infant population after thoracotomy (24).
IV. Bronchi and lungs
A. Background and development. The tracheal bud that develops from the respiratory diverticulum during the 4th week soon divides into the primary bronchial buds. These buds are the rudiments of the two lungs. By the beginning of the 5th week of gestation, each of these buds enlarges to form the primordia of the right and left main bronchi. Initially the level of the tracheal bifurcation is in the high cervical region. After approximately 4 weeks it descends to the thoracic level, and at birth, it is at the level of the fourth or fifth thoracic vertebra. Early in the 5th week, the third round of branching occurs and yields three secondary bronchial buds on the right and two on the left. These buds eventually give rise to the three lobes of the right lung and the two lobes of the left. During the 6th week, the secondary bronchi divide to form 10 tertiary or segmental bronchi in the right lung and eight or nine in the left lung (Fig. 21.6). These become the bronchopulmonary segments of the adult lung. By 16 weeks, after approximately 14 more branchings, the terminal bronchioles are produced. By 24 weeks, approximately 17 orders of branching have occurred and the respiratory bronchioles have developed. An additional seven subdivisions form during postnatal life.
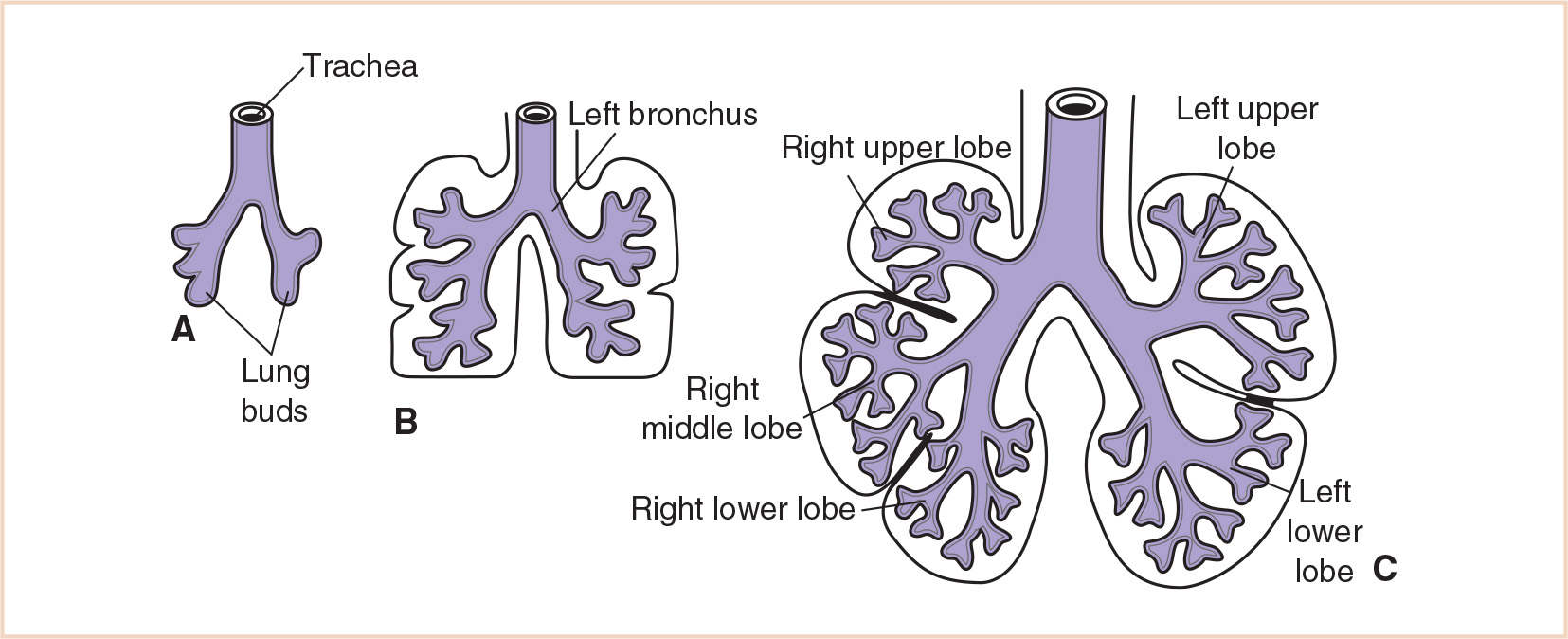
Cartilage appears in the trachea and bronchi at approximately 10 weeks gestation and in the segmental bronchi at approximately 16 weeks. Bronchial smooth muscle and cartilage are derived from the surrounding splanchnic mesoderm, which also gives rise to the vasculature of the lung and the visceral pleura. The somatic mesoderm, which lines the inside of the body wall, becomes the parietal pleura. The space between the parietal and visceral pleura is the pleural cavity.
B. Periods of lung maturation
1. Pseudoglandular period (6 to 16 weeks). Branching has continued and resulted in the formation of the terminal bronchioles. No respiratory bronchioles or alveoli are present; therefore, respiratory function is not possible.
2. Canalicular period (16 to 26 weeks). The terminal bronchioles have now divided into several respiratory bronchioles, each of which in turn divides into three to six alveolar ducts. Each duct ends in a terminal sac (primitive alveolus). Limited respiration is possible at the end of the canalicular period because some terminal sacs have become thin-walled and their surrounding tissue is well vascularized.
3. Terminal saccular period (26 weeks to birth). Many more terminal sacs develop during this period and their epithelium becomes very thin. The epithelium initially consists of cuboidal cells, but these begin to narrow into squamous cells at approximately 26 weeks. The epithelial cells establish close contact with the endothelial cells of capillaries and adequate gas exchange is now possible. The epithelial cells and mucosal lining of the bronchi are derived from endoderm.
By 26 weeks, the terminal sacs are mainly lined by squamous epithelial cells, known as type I alveolar cells or pneumocytes, which are responsible for gas exchange. Type II alveolar cells also develop and these cells produce surfactant, which lowers the surface tension at the air–alveolar interface.
4. Alveolar period (32 weeks to 8 years). By 32 weeks, structures analogous to alveoli are present. The epithelial lining of the terminal sac becomes an extremely thin squamous epithelial layer. The lungs are capable of respiration by the late fetal period because the alveolocapillary membrane is thin enough for gas exchange to occur.
Characteristic mature alveoli do not form until after birth. Before birth, the immature alveoli appear as small protuberances from the walls of the alveolar ducts. After birth, the primitive alveoli enlarge as the lungs expand. Although the alveoli increase somewhat in size, growth of the lungs is primarily due to an increase in the number of respiratory bronchioles and primordial alveoli. During the first few months of life, there is an exponential increase in the surface of the air–blood barrier, which is accomplished by the proliferation of alveoli and capillaries. Only one sixth of the adult number of alveoli (~300 million in total) is present at birth. The remaining alveoli are formed during the first few years of postnatal life through the continuous formation of new primitive alveoli. Most of this process is completed by 2 to 4 years of age, although it continues until approximately 8 years of age (26).
C. Congenital lobar emphysema (CLE)
1. Background. CLE refers to the postnatal abnormal overdistension of an otherwise anatomically normal lobe of the lung. This condition is characterized by expiratory air trapping within the lobe, which produces compression and atelectasis of adjacent lobes. If it is severe, mediastinal shift and impaired venous return can occur. Impaired gas exchange is common in CLE.
2. Incidence. CLE is rare, with a 2:1 or 3:1 male preponderance. Congenital heart disease or abnormalities of the great vessels occur in approximately 15% of infants. In particular, patients with CLE have an increased incidence of ventricular septal defect and patent ductus arteriosus (27,28).
3. Embryology/anatomy. CLE classically develops when the cartilage of the affected bronchus fails to develop, resulting in focal bronchial collapse and air trapping on expiration. However, other causes of bronchial obstruction, either intrinsic or extrinsic, may be responsible for the development of CLE. Intrinsic causes include bronchial stenosis, bronchial cysts, or redundant bronchial mucosa. Extrinsic bronchial compression may be due to abnormal vessels (such as an aberrant or enlarged pulmonary artery or ductus arteriosus), mediastinal lymphadenopathy, or mediastinal cysts or tumors. Polyalveolar lobe syndrome has also been identified as a cause of CLE. In this syndrome, the involved lobe has three to five times the normal number of alveoli present and air trapping occurs postnatally within these many alveoli.
Usually only one lobe is involved, with 40% to 50% of cases affecting the left upper lobe, 30% to 40% affecting the right middle lobe, and 20% affecting the right upper lobe. The lower lobes are affected in only 1% of cases. Occasionally more than one lobe is involved.
4. Clinical features. CLE usually presents in full term infants between the newborn period and 6 months of age, with approximately half of all patients presenting within the first 2 days of life. The clinical presentation is variable, depending on the degree of bronchial obstruction and the time it takes for overexpansion to occur. Many infants present with tachycardia, tachypnea, and retractions. If hypoxemia ensues, the infant may become increasingly agitated. Other symptoms include cyanosis, grunting, and coughing. Physical examination may reveal asymmetric chest expansion with focal hyperresonance and diminished breath sounds over the affected lobe. The diagnosis is confirmed by plain chest radiograph with typical findings including hyperinflation of the involved lobe, atelectasis of adjacent lung, mediastinal shift, and flattening of the ipsilateral diaphragm (29).
D. Pulmonary sequestration
1. Background. Pulmonary sequestration is a type of bronchopulmonary-foregut malformation that results in an abnormal, nonfunctioning mass of lung tissue lacking connection to the tracheobronchial tree. The sequestered mass receives its blood supply from one or more anomalous systemic arteries. Sequestrations can be intralobar (90% of cases) or extralobar (10% of cases). An intralobar sequestration is found within the visceral pleura of the normal lung, whereas an extralobar sequestration is located outside the lung encased within its own visceral pleura.
2. Incidence. Pulmonary sequestrations are rare lesions, comprising approximately 6% of all congenital pulmonary malformations. The incidence of intralobar sequestrations is equal in males and females, whereas extralobar sequestrations have a 4:1 male predominance. Extralobar sequestrations are associated with other congenital anomalies in up to 40% to 60% of patients. The most common association is with congenital diaphragmatic hernia (CDH), which occurs on the ipsilateral side. Others include chest wall deformities (pectus excavatum), vertebral defects, congenital heart disease, and other lung anomalies (CCAM, bronchogenic cysts). In contrast, infants with intralobar sequestrations are otherwise generally healthy and do not have any associated congenital anomalies.
3. Embryology/anatomy. There are conflicting theories on the origin of pulmonary sequestration. The most widely accepted theory holds that a sequestration occurs as a result of an accessory lung bud developing from the ventral aspect of the primitive foregut. The accessory lung bud migrates caudally with the foregut. It receives its blood supply from vessels that connect to the aorta and supply the primitive foregut. These attachments to the aorta persist to form the systemic arterial supply of the sequestration. If the bud develops before the development of the visceral pleura, it is included within the pleura and an intralobar sequestration arises. If the accessory lung bud develops after the development of the pleura, it is not incorporated within the lung visceral pleura and an extralobar sequestration results.
Intralobar sequestrations lie almost exclusively within one of the lower lobes and nearly two-thirds are on the left side. Their arterial supply is systemic in origin and comes mainly from branches of the descending thoracic aorta or less commonly the abdominal aorta. In the case of the latter, the vessels penetrate the diaphragm to supply the sequestration. Venous drainage usually occurs through one of the pulmonary veins. Foregut communication is very rare. Extralobar sequestrations can be found anywhere within the thorax, but the most common site is between the left lower lobe and the diaphragm (nearly all occur on the left side). Up to 10% of lesions are found within the abdomen (subdiaphragmatic). The systemic arterial blood supply is derived mostly from the thoracic aorta or from the abdominal aorta, with an infradiaphragmatic source in 20% of cases. Venous drainage is typically through the azygous vein. Because of the foregut derivation, communication with the esophagus or stomach may occasionally occur.
4. Clinical features. Infants born with pulmonary sequestrations are generally not symptomatic at birth because these lesions are relatively small compared to the normal lung. Intralobar sequestrations typically present as recurrent bouts of pneumonia involving the same lower lobe. Infection results from inadequate tracheobronchial drainage either from the lesion itself or from adjacent atelectatic lung. Because this process takes time to evolve, most patients present after 2 years of age. Extralobar sequestrations are usually diagnosed within the first few months or years of life. They are often discovered incidentally during the repair of a CDH (~10% to 15% of infants with CDH have one or more extralobar sequestrations) or during imaging studies for surgery or for associated congenital anomalies. Infants may have feeding difficulties if communication with the gastrointestinal tract is present. In both types of sequestration, the systemic arterial supply can result in high-output cardiac failure from shunting through the sequestration.
E. Congenital cystic adenomatoid malformation (CCAM)
1. Background. CCAM is a lung lesion that develops from adenomatous overgrowth of terminal bronchioles without concomitant alveolar growth. These lesions can be cystic, solid, or mixed intrapulmonary masses that are characterized by an absence of bronchial cartilage and bronchial glands. Communication with the tracheobronchial tree is usually maintained, and vascular supply and venous drainage are derived from the pulmonary circulation. Massive enlargement of the affected lobe may occur, causing compression and displacement of adjacent lung and other thoracic structures.
2. Incidence. CCAMs comprise approximately 25% of all congenital lung malformations. A review of 48 cases in Canada led to an estimated incidence of 1:25,000 to 1:35,000 (30). Approximately 20% of patients with CCAM have an associated congenital anomaly, with renal agenesis and cardiac defects being the most common. An association with pulmonary sequestration has also been reported.
3. Embryology/anatomy. CCAM is thought to result from focal arrest in fetal lung development sometime between the 7th and 35th week of gestation. In particular, it has been suggested that disordered development of the lobar lung bud occurs between 16 and 20 weeks, giving rise to a lesion comprising multiple interconnected cysts that are disorganized and varied in size (29). There is an increase in terminal bronchioles with no associated alveoli, bronchial cartilage, or glands.
CCAMs have no predilection for either lung, but tend to occur slightly more commonly in the upper lobes. Unilobar involvement is most common, but multilobar disease can be seen as well. Fortunately, bilateral lesions rarely occur so that when multiple lobes are involved, surgical resection can be accomplished by pneumonectomy when necessary. On rare occasions, segments of both lungs, or the entire lung, may be involved. When the lesions are large, pulmonary growth may be compromised leading to pulmonary hypoplasia.
4. Clinical features. The clinical presentation of CCAMs can be variable depending on the size of the lesions. However, 80% of affected neonates present with some degree of respiratory distress secondary to pulmonary compression or hypoplasia. Because these lesions communicate with the tracheobronchial tree, they may become overdistended due to air trapping. Other causes of respiratory insufficiency are mediastinal shift and spontaneous pneumothoraces. Typical signs and symptoms of the neonate with CCAM include tachypnea, grunting, retractions, accessory muscle use, cyanosis, and failure to thrive. In severely affected infants, the presentation may resemble that of CDH. These infants may require conventional mechanical ventilation, high-frequency ventilation, or even extracorporeal life support in extreme cases. As in CDH, prognosis depends on the amount of remaining functional lung tissue (31).
Large lesions may be associated with the development of hydrops fetalis in up to 40% of cases. Hydrops results from increased intrathoracic pressure from the lesion, which compromises venous return and leads to decreased cardiac output, high central venous pressures, and the development of effusions. Fetal demise may result. Polyhydramnios may also be seen prenatally as a result of increased intrathoracic pressure and compression of the esophagus.
If lesions are small, they may remain undiagnosed for several years or may be discovered incidentally later in life. Recurrent infection may be a presenting feature in older patients. These patients are at risk for infectious processes such as pneumonia or lung abscesses because the abnormal lung mass lacks the normal clearance mechanisms.
CLINICAL PEARL Each congenital lung lesion has several important features. CLE involves abnormal emphysematous lung that communicates with a bronchus and overexpansion is a major concern. CCAM tissue does not participate in gas exchange, but may communicate with the tracheobronchial tree (with concern for air trapping), and blood supply is usually from the pulmonary circulation. Pulmonary sequestration lesions involve nonfunctional lung tissue without bronchial connection and with blood supply from anomalous systemic arteries.







Full access? Get Clinical Tree


