Chapter 29 The acute respiratory distress syndrome (ARDS)
The acute respiratory distress syndrome (ARDS) was first described in 1967 by Ashbaugh and colleagues as the ‘acute onset of tachypnea, hypoxemia and loss of compliance after a variety of stimuli’.1 Continued research examining the underlying mechanisms and management strategies is now translating into improved outcome.
DEFINITIONS
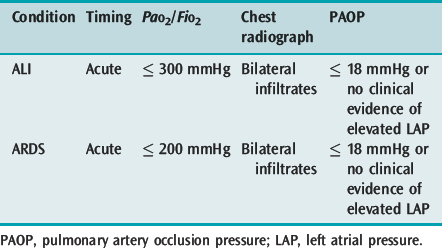
Acute lung injury (ALI) and its more severe subset ARDS describe acute hypoxaemic respiratory failure due to bilateral and diffuse alveolar damage. The most common criteria are the 1994 American–European Consensus Conference definitions2 (Table 29.1), which are broad and inclusive. However, these fail to specify an acute cause, use a PaO2/FiO2 ratio independent of respiratory support and are not specific about the radiographic criteria. The lung injury score (LIS),3 which uses a four-point score attributed to ranges of PaO2/FiO2 ratio, positive end-expiratory pressure (PEEP), respiratory system compliance and the number of quadrants involved on chest radiograph, and the Delphi definition, which demands a PaO2/FiO2 ratio ≤ 200 with 10 cmH2O PEEP, have a greater sensitivity when matched against autopsy evidence of diffuse alveolar damage.4
CHEST RADIOGRAPH AND CHEST COMPUTED TOMOGRAPHY IN ACUTE LUNG INJURY
The interpretation of the chest radiograph is central to these definitions of ALI and ARDS. However, there is considerable interobserver variability in both chest radiograph interpretation and in the definition of an infiltrate. In the American–European Consensus Conference definition the infiltrate must be bilateral and consistent with pulmonary oedema,2 whereas the LIS rates the number of quadrants with alveolar consolidation3 and the Delphi definition requires bilateral airspace disease. The intention of these descriptions is fairly clear and excludes opacity due to pleural effusion, nodules, masses, collapse and pleural thickening. However, it is desirable to improve interobserver agreement and this will require training and more specific definitions.
Chest computed tomography (CT)5 has proved extremely helpful in pathophysiological studies of ALI, has demonstrated the heterogeneity of lung inflation and is commonly used to assist clinical management. Autopsy and chest radiographs of ALI show a uniform process affecting both lungs; however, chest CT early in the course of ALI in supine patients demonstrated that there was a dorsal dependent increase in lung density, and that the ventral lung was relatively normal. In addition, CT frequently showed previously undiagnosed pneumothorax, pneumomediastinum and pleural effusion. After the second week of mechanical ventilation CT scans may demonstrate altered lung architecture and emphysematous cysts or pneumatoceles.
CT numbers or Hounsfield units can be assigned to each voxel (∼2000 alveoli in a standard 10-mm slice).5 These data can then be used to assess what proportion of a region of interest is non-aerated, poorly aerated, normally aerated or hyperinflated. Initially a single basal lung slice was studied, but it is clear that far more information can be obtained by studying the whole lung, and by using thinner slices. This allows: (1) reconstruction of the upper and lower lobes (the middle lobe is difficult to separate); (2) the same section of lung to be studied at different levels of inflation or PEEP (the lung also moves in a cephalocaudad direction with respiration); and (3) a broader picture of the lung to be obtained (lung damage is heterogeneous in ALI). However, whole-lung CT demands considerable exposure to ionising radiation, and different information, perhaps more pertinent to mechanical ventilation, is obtained from dynamic CT.
Clinical assessment of chest CT is discussed in Chapter 35, and CT findings in ALI are discussed below, in the section on clinical management.
EPIDEMIOLOGY
Estimates of the incidence and outcome from ALI and ARDS vary widely. In part this has been due to differences in the definitions used, but it also appears likely that case-mix and local factors influence outcome and incidence. Using the 1994 consensus definition the Australian incidence is 34 per 100 000 for ALI and 28 per 100 000 for ARDS;6 recent US estimates are 79 and 59 per 100 000 respectively7 – both much greater than many previous estimates. The Australian data equate to 1 in 10 non-cardiothoracic intensive care unit (ICU) patients developing ARDS, which reflects the tendency for clinicians to underestimate the incidence of ALI and ARDS.
Reported mortality rates are also influenced by the definitions used. For many years the mortality for ARDS was reported to be ∼60%; the Australian multicentre data reported mortality rates of 32% for ALI and 34% for ARDS,6 with US mortality rates a little higher at 38.5% and 41%.7 However, particular diagnostic groups such as multiple trauma have a lower mortality rate than other causes of ARDS, and patients with ALI who have chronic liver disease, non-pulmonary organ dysfunction, sepsis or age greater than 70 years (hazard ratio 2.5)8 have a higher risk of death. Consequently many factors need to be considered when assessing outcome prediction.
PULMONARY FUNCTION IN SURVIVORS
Respiratory function is most abnormal soon after discontinuation of mechanical ventilation, but usually returns towards normal by 6–12 months. Although a variety of abnormal pulmonary function tests may be found, impaired diffusing capacity is the most common. This is rarely symptomatic, but occasional patients have severe restrictive disease, and this is correlated with their cumulative LIS.9
QUALITY OF LIFE IN SURVIVORS
Compared with disease-matched ICU patients who do not develop ARDS, patients with ARDS have a more severe reduction in both pulmonary and general health-related quality of life.10 Many patients have reduction in exercise tolerance that may be attributable to associated critical-illness neuropathy and myopathy; nerve entrapment syndromes and heterotopic calcification play a role in a minority.7 Depression, anxiety and posttraumatic stress disorder are also common (20–50% of survivors).7 However, it is unclear whether neuropsychological disability is a direct consequence of the illness, or related to the associated stress. Finally, most survivors have cognitive impairments such as slowed mental processing, or impaired memory or concentration, and these correlate with the period and severity of desaturation < 90%.11 Although these data suggest that ARDS confers a specific risk of impaired quality of life, the mechanism is unclear. However, they do caution against permissive hypoxaemia as a strategy to reduce ventilator-induced lung injury (VILI).
PATIENTS AT-RISK FOR ALI AND ARDS
Clinical risk factors for the development of ALI and ARDS can be classified as either direct or indirect (Table 29.2). These identify over 80% of patients who develop ARDS; the most common risk factors are sepsis, pneumonia and aspiration of gastric contents. Multiple risk factors, low pH, chronic alcohol abuse or chronic lung disease substantially increases the incidence of ALI in at-risk patients.
Table 29.2 Clinical risk factors for acute lung injury (ALI) and acute respiratory distress syndrome (ARDS)
Rights were not granted to include this table in electronic media. Please refer to the printed book.
BIOLOGICAL MARKERS AS PREDICTORS OF ALI
In addition to identifying these clinical risk groups, there has been considerable interest in identifying possible biological markers that could be used to predict the development of ALI. Although greater specificity may be gained from sampling the epithelial lining fluid (e.g. using bronchoalveolar lavage fluid), an ideal biological marker would be more simply sampled, such as plasma. Numerous proteins, such as the cytokines interleukin (IL)-1, tumour necrosis factor-α (TNF-α) and IL-10, and von Willebrand’s factor antigen are elevated in at-risk patients and patients with ALI and ARDS; however, they are not predictive. In relatively small studies both ferritin12 and surfactant protein B13 have been shown to be predictive of ARDS. Although ferritin likely represents a non-specific oxygen free-radical response, the leakage of surfactant protein B from the alveolus into the blood is lung-specific.
PATHOGENESIS
Although it is well accepted that diffuse alveolar damage with: (1) pulmonary oedema due to damage of the alveolocapillary barrier; (2) a complex inflammatory infiltrate; and (3) surfactant dysfunction are essential components of ALI, the sequence of events is uncertain and probably depends upon the precipitating insult and host response. For example, in endotoxin-induced lung injury, hypoxaemia and reduced lung compliance occur well before recruitment of neutrophils or an increase in lung weight due to an increase in permeability.14 In addition, surfactant turnover is dramatically increased prior to these changes often thought to be typical of early ALI. Further, epithelial lining fluid sampled immediately following intubation in patients with ALI has markedly increased concentrations of type III procollagen peptide, suggestive of fibrosing alveolitis extremely early in the course of lung damage.
THE ALVEOLOCAPILLARY BARRIER
The normal lung consists of 300 million alveoli with alveolar gas separated from the pulmonary microcirculation by the extremely thin alveolocapillary barrier (0.1–0.2 μm thick). Since the endothelial pore size is 6.5–7.5 nm, and the epithelial pore size is almost one-tenth that, at 0.5–0.9 nm, the epithelium is the major barrier to protein flux.15 The surface area of the alveoli is estimated to be 50–100 m2, which is made up predominantly of alveolar type I cells, with the metabolically active type II cells accounting for ∼10% of the surface area. In turn, these cells are covered by the epithelial lining fluid, with an estimated volume of 20 ml, ∼10% of which is surfactant with the remained filtered plasma water and low-molecular-weight proteins, and a small number of cells, mainly alveolar macrophages and lymphocytes.
NEUTROPHILS IN ACUTE LUNG INJURY
Neutrophils are the most abundant cell type found in both the epithelial lining fluid (e.g. bronchoalveolar lavage fluid) and alveoli in histological specimens from early in the course of ALI. Although neutrophil migration across the endothelium or epithelium does not cause injury, when activated they release reactive oxygen species, cytokines, eicosanoids and a variety of proteases that may make an important contribution to tissue damage in ALI. Following bone marrow demargination, activated neutrophils adhere to the endothelium on their passage to the alveolus, and this may be accompanied by an early, transient leukopenia. Although neutrophils have an important role in host defence due to their bactericidal activity, there is a marked (50–1000-fold) increase in the release of cytotoxic compounds when they are activated by adherence to the endothelium, epithelium or contact with interstitial extracellular matrix proteins.16 The factors involved in adhesion of neutrophils are complex and involve the integrin family of proteins, selectins and a number of adhesion molecules.
In models of ALI, antibodies to adhesion molecules (e.g. CD11b/CD18 antibodies) ameliorate lung injury, suggesting a crucial and central role of this cell type. However, ALI occurs in neutropenic patients, and was not more common when granulocyte colony-stimulating factor was administered to patients with pneumonia.17 Clearly, other cell types play an important role, and neutrophil chemoattractants such as IL-8 must be present in the lung prior to neutrophil accumulation.
OTHER CELL TYPES INVOLVED IN ACUTE LUNG INJURY
Pulmonary endothelial cells, platelets, interstitial and alveolar macrophages and alveolar type II cells also play important roles in alveolar inflammation. Pulmonary endothelial cells express a variety of adhesion molecules and cyclooxygenase-2 (COX-2); secrete endothelin and cytokines, including IL-8;18 stimulate procoagulant activity; and ‘cross-talk’ with the alveolar macrophages and type II cells. They will be involved in generalised endothelial activation, and are subject to mechanical stress, secondary to both vascular pressure and to their close association with the alveolus. Von Willebrand’s factor antigen is synthesised by vascular endothelial cells, and although this may explain its lack of specificity for ALI, its plasma levels are a good marker of endothelial injury.19
Microvascular thrombosis is common in ALI, and contributes to pulmonary hypertension and wasted ventilation. Although platelet aggregation may contribute to ALI through release of thromboxane A2, serotonin, lysosomal enzymes and platelet-activating factor, they are less important than the other cell types.
Alveolar macrophages are the most common cell type normally found in bronchoalveolar lavage fluid, and, together with interstitial macrophages, play an important role in host defence and modulation of fibrosis. They are capable of releasing IL-6 and a host of mediators, similar to the activated neutrophil, including TNF-α and IL-8 in response to stretch,20 and may amplify lung injury. However, depletion of alveolar macrophages does not reduce neutrophil recruitment or outcome from tracheal instillation of Pseudomonas aeruginosa,21 questioning the central role of this cell type. Macrophages also release a number of factors such as transforming growth factor-α and platelet-derived growth factor that stimulate fibroblast proliferation, deposition of collagen and glycosaminoglycans, angiogenesis and lung fibrosis.
Alveolar epithelial type II cells are extremely metabolically active; they manufacture and release surfactant, control alveolar water clearance using ion pumps, express cytokines which in turn interact with surfactant production and are the progenitor of type I cells following injury. In response to both stretch and endotoxin, type II cells express IL-8 and TNF-α, with the latter cytokine augmenting Na+, and hence water, egress from the alveolus.22
CHEMOKINES IN ACUTE LUNG INJURY
The expression and secretion of chemokines (chemoattractant cytokines) at sites of inflammation are probably key proximal steps in initiating the inflammatory cascade. IL-8 appears particularly important in initiating ALI because of its ability to induce chemotaxis and activation of neutrophils. IL-8 is elevated in ALI bronchoalveolar lavage fluid within hours of the initiating insult and before recruitment of neutrophils, and in a manner that reflects subsequent morbidity and mortality. In animal models of sepsis and acid aspiration, instillation of antibodies against IL-8 prevents the recruitment of neutrophils and protects the lung. Indeed, the recruitment and retention of neutrophils require the generation and maintenance of a localised chemotactic/haptotactic gradient.23
Mediators in acute lung injury
Although many of the over 40 biologically active cytokines are implicated in ALI, TNF-α, IL-1β, IL-6 and IL-8 are the most important. However, even greater increases are found in their cognate receptors or antagonists, such as the counterregulatory cytokine IL-10, so that their biological impact is markedly reduced.24 Despite numerous studies, measurement of cytokines in blood or epithelial lining fluid has not proven predictive of the development of ALI or of mortality.
CLINICAL MANAGEMENT
MECHANICAL VENTILATION
Patients present with acute hypoxaemic respiratory failure, and an increase in the work of breathing, usually requiring mechanical ventilation (Table 29.3). The role of non-invasive ventilation in ALI is uncertain as there appears to be a greater complication rate, perhaps due to delayed intubation.25 However, non-invasive ventilation is worth considering in particular circumstances (see Chapter 33).
Table 29.3 Pathophysiology of acute lung injury (ALI) and adult respiratory distress syndrome (ARDS)
| Feature | Cause(s) |
|---|---|
| Hypoxaemia | True shunt (perfusion of non-ventilated airspaces) |
| Impaired hypoxic pulmonary vasoconstriction | |
| V/Q mismatch is a minor component | |
| ↑ Dependent densities (CT) | Surfactant dysfunction alveolar instability |
| (Collapse/consolidation) | Exaggeration of normal compression of dependent lung due to ↑ weight (↑ lung water, inflammation) |
| ↑ Elastance (↓ compliance) | Surfactant dysfunction (↑ specific elastance) |
| ↓ Lung volume (‘baby lung’) | |
| ↑ Chest wall elastance | |
| Fibrosing alveolitis (late) | |
| ↑ Minute volume requirement | ↑ Alveolar dead space (VDphys/Vt often 0.4–0.7) |
| ↑ VCO2 | |
| ↑ Work of breathing | ↑ Elastance |
| ↑ Minute volume requirement | |
| Pulmonary hypertension | Pulmonary vasoconstriction (thromboxane A2, endothelin) |
| Pulmonary microvascular thrombosis | |
| Fibrosing alveolitis | |
| Positive end-expiratory pressure |
The method and delivery of ventilatory support must take into account the pathophysiology of ALI and ARDS. For many years laboratory studies have described VILI, and the most important clinical study was the ARDS Network study where 861 ALI patients from 75 ICUs were randomised to receive a tidal volume (VT) of either 12 or 6 ml/kg predicted body weight.26 Mortality was reduced by 22% from 40% to 31% in the lower-VT group. There was a strict PEEP and FiO2 protocol, and patients were ventilated with assist-control ventilation to avoid excessive spontaneous VT. Meta-regression of five clinical studies trialling different VT strategies found that a VT < 7.7 ml/kg predicted body weight (pbw) was protective, and that above 11.2 ml/kg pbw was borderline detrimental.27 Whether this protective effect of lower VT is due to lower static distending pressure of the respiratory system is contentious; however, Hager and colleagues28 found that lower VT ventilation was protective across all quartiles of plateau pressure (Pplat) in the ARDS Network trial, and that no safe upper limit of Pplat could be identified.




Full access? Get Clinical Tree


