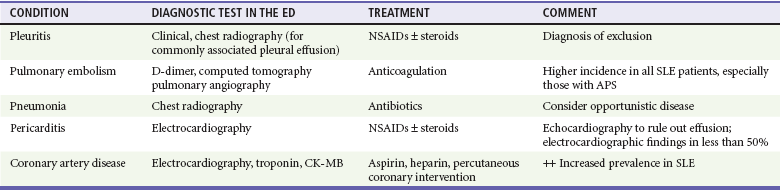
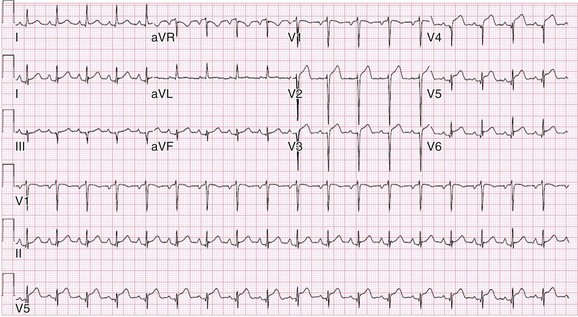
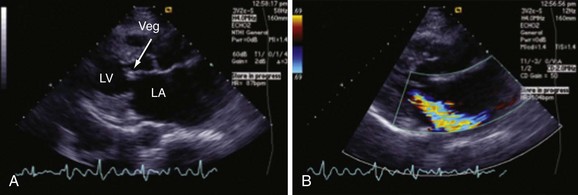
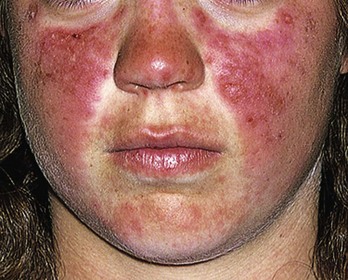
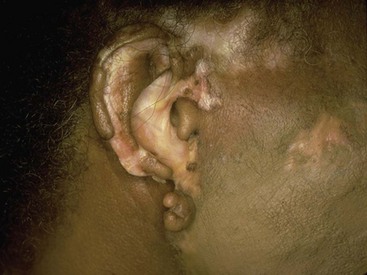
Chapter 118 Systemic lupus erythematosus (SLE) is a multisystem, autoimmune disorder that poses a challenge to the emergency physician with its protean and occasionally dangerous manifestations. Morbidity for SLE patients is typically mediated through organ inflammation and destruction or the consequences of therapeutic immunosuppression. Further complicating lupus is the frequent association with the antiphospholipid syndrome (APS) and its corresponding venous and arterial thromboses. The diagnostic criteria reflective of its complexity and immunologic basis were first described in 1971.1 Today, the emergency department (ED) is a common venue for the evaluation and treatment of patients suffering from SLE.2 SLE is present in 20 to 70 per 100,000 of the general population.3 Although great variation in incidence exists worldwide, it is clear that two groups have consistently higher incidences: women, representing 90% of cases; and African Americans.3 Dramatic rises in incidence of SLE have been described during the past several decades, but it is suspected that this may be due to the implementation of more sensitive diagnostic criteria.4 With women representing 90% of SLE cases,5 a strong role for estrogen in disease development has been well supported in large cohort studies.6–8 The disease also has a strong genetic link, with high rates of monozygotic twin concordance and tremendous genetic overlap at the human leukocyte antigen (HLA) alleles.9 SLE may cause disease in nearly any organ system in the body through inappropriate immune response to self and is often described as the prototype of all systemic, autoimmune disorders. Like its cause, its exact pathophysiologic mechanism is not completely elucidated. When a cell cycle ends (apoptosis), intracellular contents, including DNA-rich nucleosomes, are released into the bloodstream. For patients with SLE, this cellular debris may be regarded as nonself (i.e., an antigen) by the immune system. After exposure to these self antigens, the familiar immunologic cascade of antigen-presenting cells, T cells, and ultimately B cells and their plasma cell progeny is exacted, and patients form autoantibodies against their own cellular contents.10 These autoantibodies (which include anti–double-stranded DNA antibody, a commonly ordered rheumatologic assay in evaluation for SLE) may then mediate organ-specific disease and inflammation. This may occur through either direct attack and injury to parenchymal tissue or other mechanisms,11 such as formation of immune complexes that are deposited in tissue.12 On the basis of this immunologic understanding of the pathogenesis of SLE, the rationale for immunosuppression as both chronic and acute therapy for SLE can be appreciated. Although some elements, such as the malar rash, that form the diagnosis of SLE are nearly pathognomonic for SLE, a confident initial diagnosis in the ED may otherwise be rare or difficult. The literature is replete with ED-based case reports of extreme initial presentations with conditions such as Guillain-Barré, cardiac tamponade, and fulminant renal failure that subsequently were confirmed as complications of unrecognized SLE13–17; however, in all cases the diagnosis of SLE was applied retrospectively. Because the workup for SLE is commonly conducted in a series of outpatient visits and relies on laboratory investigations that are uncommonly carried out in the ED, establishment of a new diagnosis of SLE in the ED is difficult. If a new diagnosis of SLE is suspected in an otherwise well patient, referral for an expedited workup is reasonable in most circumstances. The American College of Rheumatology classification criteria for SLE are presented in Table 118-1.18,19 Table 118-1 American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus18,19 *Patients exhibiting at least 4 of the 11 conditions listed here at any point in time meet the criteria for diagnosis of SLE. Emergency physicians are commonly faced with evaluating the acute complaints of patients with an established diagnosis of SLE.2 The workup of SLE-related complications is based on use of a disease-specific differential diagnosis. Even with good medication compliance, SLE patients are prone to exacerbations of their disease, typified by worsening physical symptoms from increased organ inflammation and destruction. These exacerbations may involve organs already affected by SLE or new conditions manifested in previously unaffected organ systems. As a systemic disease, worsening symptoms in one organ system should reasonably prompt evaluation for progressive disease activity in other organs. True to the broad scope of SLE, the most accepted scoring system to grade SLE exacerbations accounts for 20 different markers of disease activity across 9 organ systems.20 Although such scoring systems have little role in the ED, it is important to identify when overall disease activity is increased as it typically signals the need to initiate or to escalate systemic therapy. In many cases, patients themselves are able to provide direction about the predictable course of their exacerbations and can be helpful in decision-making for therapy and disposition. Fever.: Improving survival for patients with SLE during the past several decades has been achieved with the use of improved immunosuppressive regimens. As a result, patients with SLE remain commonly affected by infections mediated by both typical and opportunistic organisms, suffering higher than average death rates from these infections.21 In one case series, infection was the leading reason for SLE patients to be admitted to the intensive care unit, superseding both renal failure and cardiovascular disease.22 Whereas absence of fever is insufficient in many cases to rule out infection, the presence of a fever is grounds for concern. Fever may also be due to increased overall disease activity.23,24 Given the heightened risk for SLE patients with infection, determination that a fever is due strictly to disease exacerbation should be made cautiously.21 The majority of infections in SLE are due to skin, lung, or urinary sources and are caused by typical organisms. However, largely because of the immunomodulating therapies that form the cornerstone of SLE management, opportunistic diseases are possible; Pneumocystis (carinii) jiroveci pneumonia, cryptococcal meningitis, Listeria infection, and herpes zoster have all been described.25 Neuropsychiatric Presentations.: According to the American College of Rheumatology, there are 19 different clinical manifestations of neuropsychiatric SLE, some of which are included in the diagnostic criteria (see Table 118-1).26 These 19 different presentations range vastly and include seizures, confusion, cranial neuropathies, demyelinating syndromes, myasthenia gravis, depression, psychosis, and anxiety. It may not be possible in the ED to determine whether such manifestations are due to neuropsychiatric SLE or to other, independent pathologic processes. Headache.: In general, headaches in a patient with SLE may be evaluated in a fashion similar to that for headaches in the general population. In one prospective cohort study, for instance, it was found that primary headache disorders, such as migraine and tension headaches, occur with the same frequency in SLE patients as in the general population.27 This was further supported by an extensive meta-analysis examining the implications of headache in SLE patients.28 In this study, the authors found no relationship between headache and SLE disease activity. Thus, despite recommendations to consider headache as suggestive of neuropsychiatric SLE or so-called lupus cerebritis,26 isolated headaches in the context of lupus may be treated in a fashion typical of other primary headache disorders. When a more malignant cause of headache is suspected, as suggested by meningismus, fever, or focal neurologic findings, an aggressive workup is warranted. When concern exists for an infectious cause of headache, consideration for opportunistic infections, such as cryptococcal meningitis, should be made in an immunosuppressed SLE patient.29 Further, because of common comorbid APS, consider sinus thrombosis in the SLE patient with new-onset focal central nervous system findings and headache.30 Seizure.: Because of incompletely understood mechanisms, seizures may occur as result of SLE disease activity. In one large series, seizures were observed in 11% of SLE patients, and in the majority of cases, either stroke or comorbid APS was present.31 Because of the high incidence of seizures, nonconvulsive status epilepticus is a consideration in SLE patients with an acute onset of altered mental status. The workup and management of seizures do not differ between patients with SLE and the general population. Focal Neurologic Findings.: On occasion, the patient with SLE may present with focal motor weakness. In those with SLE who also have APS, there is a significantly increased risk of stroke related to spontaneous arterial thrombosis.32 Demyelinating disease, such Guillain-Barré and cranial neuropathies, have also been occasionally attributed to SLE from unclear mechanisms. Psychiatric Symptoms.: Mood disorders, psychosis, and anxiety are considered potential manifestations of neuropsychiatric SLE.26 Distinguishing SLE-related from functional causes of psychiatric presentations poses a challenge and may not be possible in the ED. Although psychiatric presentations of SLE in the ED have not been directly studied, in the absence of concern for an organic cause of altered mental status, patients with SLE are approached in the same manner as patients without SLE. Corroboration with the patient’s rheumatologist may provide insight to history of similar events or guidance for treatment. Cardiorespiratory Presentations and Diseases.: Chest pain, in the form of either pleuritis or pericarditis, is among the diagnostic criteria for SLE (see Table 118-1). In addition to pericarditis and pleuritis, coronary artery disease (CAD), pulmonary embolism, and musculoskeletal causes are also common. Other forms of cardiac disease that occur without chest pain, such as verrucous (Libman-Sacks) endocarditis and various arrhythmias, may also be present.15 Coronary Artery Disease.: There is a dramatically increased risk of CAD in the context of SLE. This was demonstrated in a compelling retrospective cohort study of 500 women with lupus compared with age-matched controls from the original Framingham data. In this study, a 52-fold increased risk of myocardial infarction was determined in women between the ages of 35 and 44 years.33 These findings of increased risk of CAD have been supported by a number of other prospective trials. In one series, patients with SLE were found to be five times as likely to have CAD (based on coronary artery calcium scoring) as patients without SLE.34 In another series that profiled an even younger subset, it was found that among otherwise low- to moderate-risk women aged 22 to 45 years with complaints of chest pain, dyspnea, or decreased exercise capacity, there was an alarming 82% prevalence of CAD as determined by nuclear imaging. There was a similarly concerning prevalence of 43% of CAD in the asymptomatic “control” group who also had SLE.35 Further, traditional cardiac risk factors are found less often in patients with SLE who suffer consequences of CAD, suggesting that CAD in patients with SLE cannot be explained by the higher incidence of hypertension or hypercholesterolemia that these patients often exhibit.36 Thus, whereas there are many considerations for the cause of chest pain in patients with SLE (Table 118-2), CAD must be considered highly, and in fact more highly in patients who would otherwise be deemed to be low risk (young, reproductive age women). It is therefore appropriate to have a low threshold to initiate a workup with electrocardiography, cardiac biomarkers, and stress testing to rule out acute coronary syndromes in patients with chest pain and SLE. In addition to being at risk for development of CAD, patients with SLE who are treated with percutaneous coronary intervention appear to fare poorly. In one retrospective analysis of 28 patients with SLE receiving percutaneous coronary intervention, reintervention, death, and recurrent myocardial infarction were significantly more likely compared with a similar cohort without SLE.37 Pericardial Disease.: Pericardial effusion and pericarditis occur commonly in patients with SLE. It is estimated that symptomatic pericarditis occurs in 25% of patients with SLE during the course of their lives and that pericardial effusion occurs even more often, frequently in a silent fashion, in up to 50%.38 In clinically evident SLE-related pericarditis, dyspnea or pleuritic chest pain is common. Typical electrocardiographic findings (Fig. 118-1) have been shown to be present in less than half of cases, however.39 Given the increased risk of CAD in patients with SLE, ST elevation of any kind found on an electrocardiogram warrants very careful consideration. When diagnostic uncertainty exists, echocardiography, cardiac enzymes, or cardiac catheterization may be complementary. Treatment of pericarditis includes nonsteroidal anti-inflammatory drugs (NSAIDs) and glucocorticoids, as discussed later. Pericardial tamponade, because of its dramatic nature, continues to receive attention as an initial presentation of SLE40; however, large case series have shown that this life-threatening presentation occurs in less than 1% of patients with lupus.41 In large effusions without tamponade, medical management with high-dose glucocorticoids (1-2 mg/kg of methylprednisone) has shown favorable outcomes in averting the need for pericardiocentesis.39 Pulmonary Embolism.: Pulmonary embolism and deep venous thrombosis occur with increased incidence in patients with SLE. This is especially true for patients with SLE who also carry antiphospholipid (aPL) antibodies. These patients have an estimated 20- to 30-fold increased risk for thrombosis compared with the general population. Patients with SLE who do not have aPL antibodies still have a sixfold greater likelihood for development of pulmonary embolism and deep venous thrombosis compared with patients without SLE.42,43 Thus, all patients with SLE, regardless of their aPL antibody status, which is often unknown on presentation, should be considered at increased risk for venous thromboembolism. Pleuritis.: Pleuritis, due to autoantibodies acting against the pleura itself, is the most common respiratory condition occurring in SLE.44 Characterized by pleuritic chest pain with or without a pleural effusion or pleural rub, it has symptoms that overlap with those of other more serious conditions (see Table 118-2). If a pleural effusion is present, analysis of pleural fluid for antinuclear antibodies may be the most accurate method to confirm diagnosis but may be impractical in the ED.45 A diagnosis of pleuritis should be arrived at only after other causes of pleuritic chest pain in these thrombophilic, immunosuppressed patients have been ruled out. Musculoskeletal Chest Pain.: Musculoskeletal chest pain, related to underlying pectoral, intercostal muscle, or costochondral joint inflammation, may also occur in SLE. Similar to pleuritis, the threat of a more malicious underlying cause of pain should prompt the clinician first to seek other causes before arriving at this more benign cause. Treatment with NSAIDs or acetaminophen is generally appropriate. Pneumonia.: Pneumonia is the third most common infection in patients with lupus, after skin and urinary tract infections.44 Choice of antimicrobial coverage is determined by the severity of the infection and the degree of immunosuppression. For patients receiving long-term glucocorticoids or cyclophosphamide, coverage for organisms such as Pseudomonas and Legionella generally requires the use of a carbapenem or a fourth-generation cephalosporin in addition to either a respiratory fluoroquinolone or macrolide. There is no evidence to support the routine coverage for P. jiroveci in these patients. Undifferentiated Dyspnea.: Mitral valve insufficiency due to noninfectious vegetations, known as Libman-Sacks lesions, may cause a patient to experience dyspnea on exertion or, in rare severe cases, pulmonary edema (Fig. 118-2). Anemia, present in up to 50% of patients with SLE, may lead to a chief complaint of dyspnea. Both anemia of chronic disease and hemolytic anemia are common; hemolytic anemia is more typical in those also carrying aPL antibodies.46 In cases of severe anemia recognized in the ED, a Coombs test can be diagnostic for immunologic hemolytic anemia. Other less common but noteworthy causes of shortness of breath in a patient with SLE include interstitial lung disease, lupus pneumonitis, diaphragmatic disease (so-called shrinking lung syndrome), and pulmonary hypertension.47 Musculoskeletal Presentations.: Arthritis commonly afflicts those with SLE, and increasing severity of joint pain may be a marker of increasing disease activity or an SLE flare.20 Arthritis or arthralgias are typically symmetrical and nonerosive (unlike rheumatoid arthritis) and may involve multiple joints. Arthritis is most commonly present in the hands, wrists, and knees but may be manifested in any joint. Aside from mild tenderness, the joints are often normal to physical examination and rarely experience deformity.48 Uncommonly, septic arthritis may complicate SLE. In one large retrospective analysis of SLE-related hospitalizations, only 0.3% of admissions were for septic arthritis.49 Possibly because of immunosuppressant use, including corticosteroids, Salmonella is an unusually frequent culprit organism that was isolated in 59% of cases in one study and most commonly found in the hip.49 An isolated swollen joint is not typical of SLE and prompts consideration for infectious arthritis. If septic arthritis is suspected, diagnostic arthrocentesis is recommended. Empirical treatment with 1 g of vancomycin and 1 g of ceftriaxone will cover likely organisms, including Salmonella. Myalgias are common in SLE and may be an early marker of increasing disease activity for some patients. Generalized muscle pain is typical, but muscle weakness is uncharacteristic.50 If muscle weakness is present, an underlying myositis or myopathy secondary to steroid use is considered. Gastrointestinal Presentations.: The most common gastrointestinal manifestation of SLE is oral ulceration, which occurs in nearly a third of patients with SLE. Treatment includes local symptom management (chlorhexidine mouthwashes, viscous lidocaine) combined with systemic therapy (hydroxychloroquine) in more severe or refractory cases.51 Abdominal pain commonly complicates SLE.52 The causes of abdominal pain, with the exception of lupus enteritis, are largely similar to those in patients without SLE, such as pancreatitis, gastroenteritis, and peptic ulcer disease.53 Lupus enteritis (also known as mesenteric vasculitis) typically results in diffuse abdominal pain and is the most common cause of acute abdominal pain in SLE.53,54 The abdominal pain itself may also be associated with nausea, vomiting, and nonbloody diarrhea.55 Laboratory investigations, such as white blood cell count, hemoglobin level, platelet count, and erythrocyte sedimentation rate (ESR), are not helpful as they do not differ significantly between those with enteritis and those with abdominal pain due to other causes.53 Although no “gold standard” for the diagnosis of lupus enteritis has been established, computed tomography (CT) is the most useful test to assess for this condition. Several CT scan findings are supportive, including bowel wall thickening (Fig. 118-3), engorgement of mesenteric vessels, and increased attenuation of mesenteric fat.56 When lupus enteritis is suspected, early pulse steroids, at a dose of 1 to 2 mg/kg of methylprednisolone per day, should be administered. Surgical consultation should be obtained in severe cases or when bowel necrosis has occurred. With aggressive medical therapy, which may also include cyclophosphamide, the need for surgical management is uncommon.53 Other, less common gastrointestinal illnesses to which SLE patients are predisposed include protein-losing enteropathy and intestinal pseudo-obstruction.57 Last, in assessing abdomen pain in patients with SLE, clinicians should be mindful of the effects of chronic steroid use and increased risk for both hollow viscus perforation and peptic ulcers, which may occur in the absence of traditional symptoms. Dermatologic Presentations.: The most characteristic cutaneous manifestation of SLE is the malar rash. The rash has a “butterfly” distribution of raised erythema over the bridge of the nose and malar eminences while sparing nose and nasal-labial folds (Fig. 118-4). The other most common skin lesion found in SLE is the discoid rash. Discoid lesions are circular and raised, scaly lesions that may be commonly found on the face, scalp, and ears, often in association with pigment change, alopecia, and severe scarring (Fig. 118-5). Cutaneous lupus may exist in isolation without systemic involvement. Mild cases of worsening cutaneous lupus are treated topically with 1% hydrocortisone cream. Moderate to severe cases are treated with topical calcineurin inhibitors or even systemic therapy (e.g., hydroxychloroquine) in consultation with the patient’s internist or rheumatologist.58 In nearly all cases of SLE, avoidance of sun exposure is advisable and will help minimize cutaneous disease. Renal Disease.: Renal disease develops in approximately a third of SLE patients.44 Given a high prevalence and morbidity of renal disease as well as its tendency to carry few if any symptoms, screening urinalysis and assessment of renal function are prudent in nearly all but the most straightforward presentations of SLE to the ED. Lupus nephritis may cause a nephrotic-type disease, primarily characterized by proteinuria (>2 g/day) or a more nephritic picture with hematuria and increased creatinine concentration.59 If worsening renal dysfunction is discovered, admission should be considered for hydration and escalation of therapy that may include both pulse steroids and cyclophosphamide. The remarkable improvements in life expectancy and disease control in SLE carry a cost. Chronic steroid use is associated with a well-known host of consequences, such as increased risk of CAD, osteoporosis, avascular necrosis, psychosis, hyperglycemia, and weight gain, among many others. More potent chemotherapeutic medications, such as cyclophosphamide, methotrexate, and azathioprine, may profoundly influence the patient’s immune response and cause increased vulnerability to conventional and opportunistic infections. Chronic NSAID use for the musculoskeletal consequences of SLE may contribute to peptic ulcer disease, especially if they are coadministered with glucocorticoids. Therapy with antimalarials, such as hydroxychloroquine, is common and generally well tolerated; however, retinopathy and both QT interval prolongation and refractory ventricular arrhythmias have been associated with prolonged use.60 In all but the most trivial presentations, order a serum creatinine concentration, urinalysis, and complete blood count because renal dysfunction, proteinuria, anemia, or thrombocytopenia may silently complicate SLE and be an important marker of increased disease activity. Leukocytosis, already a nonspecific finding, may be even less contributory in SLE patients taking glucocorticoids. When a new diagnosis of SLE is being entertained, other more specific immunologic assays, such as antinuclear antibody, anti-DNA, anti-Sm antibody, and aPL antibody, can be ordered in the ED with the intent of outpatient follow-up for evaluation of results. Of note, antinuclear antibody is present in nearly all SLE patients but is also present in 50% of people who do not have SLE. Thus, the absence of antinuclear antibody virtually excludes the diagnosis.61 Unlike in other inflammatory disorders, C-reactive protein does not typically rise in response to increased disease activity in SLE. It does, however, rise in the setting of infection and thus may be a useful laboratory marker to distinguish between a flare and infection. The ESR has less of a role as it has been shown to be elevated in SLE patients experiencing either a flare or an infection.62 Coagulation profile may show an elevated partial thromboplastin time (PTT) with a normal international normalized ratio (INR) in patients with SLE. In these cases, suspicion for comorbid APS should be raised (see later section on special considerations). Radiography.: In SLE, a chest radiograph may point to diagnoses such as pleuritis (pleural effusion); pericardial disease (enlarged cardiac silhouette); pneumonia, including atypical and opportunistic infections (airspace disease); and pulmonary embolism (normal). Computed Tomography.: CT scanning may be of particular value in identifying lupus enteritis in the patient with abdominal pain, and CT pulmonary angiography has become the preferred imaging modality for pulmonary embolism. In both cases, attention to the presence or absence of lupus nephritis must be made to guide the appropriate and safe use of intravenous contrast material. Bedside echocardiography may be carried out to evaluate the presence of pericardial fluid when pericarditis is suspected on the basis of clinical or electrocardiographic findings. A number of studies have documented the safety and accuracy of emergency physicians’ identifying pericardial effusions with point-of-care ultrasound examination.63 In keeping with its highly varied presentations and permutations of disease activity, a number of other diseases may be confused with early SLE before its proper diagnosis. These include undifferentiated connective tissue disease, primary Sjögren’s syndrome, primary APS, fibromyalgia with positive antinuclear antibody, idiopathic thrombocytopenic purpura, drug-induced lupus, and early rheumatoid arthritis.64 For those with an established diagnosis of SLE, chief complaint–driven considerations for acute presentations are listed in Table 118-3. Table 118-3 Nearly all noninfectious consequences of SLE are managed with introduction or escalation of immunomodulatory therapy. In the majority of disease states, a pulse dose of methylprednisolone at 1 to 2 mg/kg is appropriate. See Table 118-4 for additional therapeutic options for escalating disease severity. Table 118-4 Medications and Typical Dosing Range for Acute Exacerbations of SLE In cases of isolated cutaneous findings, topical treatment with corticosteroids is preferable to systemic therapy. Topical therapy may be initiated with 1% hydrocortisone cream to the affected area. In cases in which higher potency topical steroids are necessary, a preparation of 0.05% betamethasone applied once daily to the affected area for 2 weeks is appropriate.65 Present in nearly 40% of SLE patients,66 the antiphospholipid syndrome (APS) is considered when patients both have a clinical history of thrombosis and are carriers of any one of a particular set of aPL antibodies directed against three specific serum proteins.67 These antibodies include the anticardiolipin antibody, the misleadingly named lupus anticoagulant, and the anti–β2-glycoprotein I antibody. The most thrombogenic of the antibodies is the lupus anticoagulant, with an odds ratio as high as 16 for the development of venous thromboembolism and 10 for arterial thrombosis (stroke).32 In addition to being a strong risk factor for both venous and arterial thrombosis, the presence of APS in SLE has been shown to be an independent predictor of more severe disease68 and to be associated with increased intensive care unit mortality.44 Whether it is present in the context of SLE or independently, APS is an important cause of morbidity and mortality due to thrombosis.32,69 Clinical Features.: APS may present with any number of clinical features (Box 118-1), typically related to thrombosis or thromboembolism. A small subset of those with APS may present with multiple thrombotic sites and organ failures simultaneously. This condition is known as catastrophic APS.
Systemic Lupus Erythematosus and the Vasculitides
Systemic Lupus Erythematosus
Epidemiology
Principles of Disease
Anatomy and Physiology
Clinical Features
The Patient with Undiagnosed or Suspected SLE
CONDITION*
DESCRIPTION
Malar rash
Butterfly-shaped, red rash on the face that spares the nasolabial folds
Discoid rash
Erythematous raised patches with adherent keratotic scaling and follicular plugging (atrophic scarring may occur in older lesions)
Photosensitivity
Skin rash as a result of unusual reaction to sunlight, by patient history or physician observation
Oral ulcers
Oral or nasopharyngeal ulceration, usually painless, observed by a physician
Nonerosive arthritis
Two or more peripheral joints, characterized by tenderness, swelling, or effusion
Serositis
History or clinical evidence of either pericarditis or pleuritis; includes otherwise unexplained pericardial or pleural effusions
Renal disorder
Persistent proteinuria (>0.5 g/day) or cellular casts (red cell, hemoglobin, granular, tubular, or mixed)
Neurologic disorder
Otherwise unexplained seizures or psychosis
Hematologic disorder
Any of: hemolytic anemia, leukopenia, lymphopenia, or thrombocytopenia
Immunologic disorder
Positive serology for any of: anti-DNA antibody, anti-Sm antibody, or antiphospholipid antibody (including anticardiolipin antibodies, lupus anticoagulant, or false-positive serology for syphilis)
Positive antinuclear antibody
Abnormal antinuclear antibody titer at any time in the absence of medications that may cause drug-induced lupus
Acute Presentations in the Patient with Known SLE
SLE Exacerbation
Specific Symptoms and Presentations
Complications due to Medications
Diagnostic Strategies
Laboratory
Radiology
Special Tests
Differential Diagnosis
CHIEF COMPLAINT
CONDITION
Pleuritic chest pain
Pericarditis, pleuritis, pulmonary embolism, pneumonia, musculoskeletal chest wall pain
Delirium
Neuropsychiatric lupus, steroid psychosis
Leg swelling
Deep venous thrombosis, renal failure, right-sided heart failure (pulmonary embolism, pulmonary hypertension), protein-losing enteropathy
Shortness of breath
Pneumonia, anemia, pericarditis or pericardial effusion, pleuritis or pleural effusion, interstitial lung disease, shrinking lung syndrome
Pruritic or painful rash
Discoid SLE, drug reaction, sun exposure
Abdominal pain
Lupus enteritis, peptic ulcer disease, pancreatitis, pseudo-obstruction
Fever
Infection, increased disease activity
Arthritis
Arthralgias (lupus flare), osteoarthritis, septic arthritis, unrelated (gout, fibromyalgia)
Management
MEDICATION
MEDICATION CLASS
TYPICAL STARTING DOSE AND ROUTE
Methylprednisolone
Glucocorticoid
1-2 mg/kg IV, once daily
Prednisone
Glucocorticoid
1-2 mg/kg, PO, once daily
Hydroxychloroquine
Antimalarial
200-400 mg PO, once daily
Cyclophosphamide
Alkylating agent
500-750 mg/m2 IV, once
Azathioprine
Antimetabolite (purine analogue)
25-50 mg/day (IV or PO)
Dermatologic Treatment
Special Considerations

Full access? Get Clinical Tree