Chapter 19 Structure and Function of the Heart
Anatomic Development and Structure
Gross Anatomy
The basic form of the human heart and great vessels is complete 8 weeks after conception, after which the structures grow and mature. The ventricular mass enlarges by cellular hyperplasia and hypertrophy; hyperplasia previously assumed to cease after birth has recently been shown by carbon-14 dating to occur at an annual turnover rate of 1% at age 25 years, decreasing to half that value by age 75 years.1 Increase of ventricular volumes is believed to depend on the increasing flow through each ventricle; diverting flow from a ventricle is believed to cause hypoplasia of that ventricle and its associated great artery. Before birth, left and right ventricles have equal wall thickness. After birth and clamping of the umbilical cord, there is a rise in systemic vascular resistance and a decrease in pulmonary vascular resistance; as a result, the left ventricle becomes thicker than the right ventricle. Left ventricular wall thickness is proportional to the logarithm of age from conception.2 The ventricular septum is flat in the fetus. After birth, it bulges into the right ventricle and functions like part of the left ventricle. In the embryo, coronary arteries form in the embryonic epicardial tissue3 and join the aorta to supply flow to the thickening heart muscle, which can no longer get enough blood from sinusoids from the ventricular cavity.
Muscle fibers in the ventricles form a complex helical array. Fibers in the left ventricular midwall are circumferential, parallel to the atrioventricular groove. From this position the fibers twist gradually as they move toward each surface so that at the epicardial surface they are 75 degrees and at the endocardial surface 60 degrees from the circumferential fibers.4 Some investigators believe that the muscle fiber layers form one continuous sheet that is wrapped around itself like a turban.5 When the ventricle is dilated, the fiber angles change and become less effective in ejecting blood.6
Microscopic Anatomy
The myocardium is a syncytium made of branching fibers, each consisting of bundles of myocytes in series. The myocytes are joined to adjacent myocytes by the intercalated disc, a set of mechanical junctions: adherens junctions with N-cadherin, catenins, and vinculin; desmosomes with desmin, desmoplakin, desmocollin, and desmoglein; and gap junctions with connexins and N-cadherin.7–9 The gap junctions transmit the electrical impulse from one cell to the next.
Myocyte
The major components of the myocyte are the sarcomeres, which contain the myofibrillar contractile apparatus; the mitochondria, which contain enzymes for energy production; the sarcolemma, which contains the cell envelope and its extensions into the cytoplasm; the sarcoplasmic reticulum; and the cytosol. The numerous proteins in these structures not only play a role in normal function but, if abnormal for genetic or extraneous reasons, contribute to myocardial dysfunction.10
Contractile Apparatus
The functional unit is the sarcomere, defined as the structure between two transverse Z lines,11–13 representing discs that contain proteins such as α-actinin and filamin that connect the actin and titin filaments of adjacent myocytes. On each side of the Z line is a light zone, the I (isotropic) band, and in the center of the sarcomere are two dark zones, the A (anisotropic) bands, separated by a light H band in the middle of which is a dark thin M band (Figure 19-1). The I bands contain paired thin filaments of actin coiled in a helix and attached to the Z lines. In humans, cardiac α-actin makes up 80% of the actin in fetuses and neonates, while skeletal α-actin makes up 60% of the total in adults.14 Two long tropomyosin filaments lie in the grooves between each pair of actin filaments (Figure 19-2).15 Every 400 Å, near the crossover points of two actin filaments, is a troponin complex with the following three distinct troponins: (1) troponin T, which binds troponin to tropomyosin; (2) troponin I, which inhibits actin-myosin interaction; and (3) troponin C, which is a high-affinity calcium receptor. The thin actin filaments overlap with thick myosin filaments at the A bands. These myosin filaments are composed of light and heavy chains. The light chains coil around each other to form the long core of the myosin molecule. The heavy chains form globular myosin heads that project from the sides of the thick filament toward the actin molecules (see Figure 19-2). A collar of cardiac myosin-binding protein C encircles the thick filaments. Mutations of this protein are a common cause of hypertrophic cardiomyopathy.16 Between two A bands there is usually a thin lighter band, the H band, which has myosin but no actin filaments.13,15
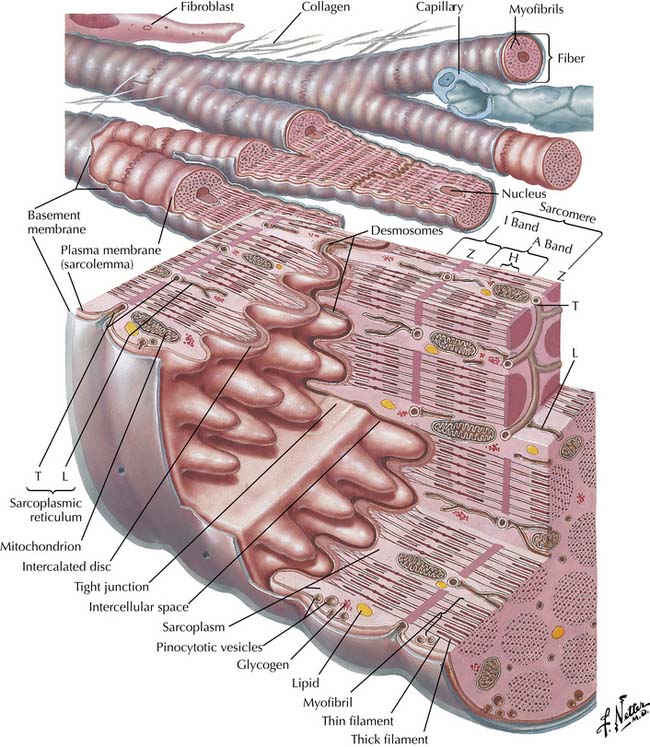
Figure 19–1 Diagram of cardiac muscle unit showing organization of structural and contractile elements.
(Netter illustration from www.netterimages.com. Elsevier Inc. All rights reserved.)
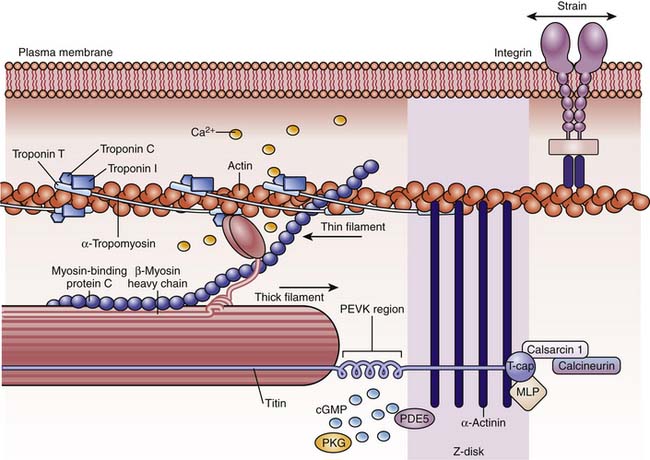
Titin, the largest known molecule (molecular weight 3-3.6 MDa, 1 mm long), is the third most abundant fibrillar protein. It extends from the Z band to the M band, has two isoforms, and is the main protein responsible for the elastic behavior of the myocyte.17 It is essential for sarcomere assembly and for sensing sarcomere length18 and, with myomesin (not shown in figure), supports the actomyosin filaments (see Figure 19-2).
Myocytes have fewer myofibrils and more water and cytoplasm before birth than after birth, and the myofibrils do not have the uniformly parallel arrays that they will have after birth.19
Sarcolemma and Sarcoplasmic Reticulum
The cell membrane contains receptors, ion channels, pumps, and exchangers. It has indentations overlying the Z bands, and from these indentations small tubules termed T (for transverse) tubules penetrate the cell. Abutting against the T-tubules are dilated expansions of the sarcoplasmic reticulum (junctional reticulum or cisternae), which join the free sarcoplasmic reticulum, a network of longitudinal tubules inside the cell that surround the thick (myosin) filaments. These tubular systems modulate the entry of calcium to, or its exclusion from, the cytoplasm.11,19
The cisternae contain the calcium-binding protein calsequestrin, whereas the longitudinal tubules contain phospholamban and the adenosine triphosphate (ATP)-dependent calcium pump.19,20 Phospholamban inhibits the affinity of the sarcoplasmic reticulum Ca2+-ATPase (SERCA) pump for calcium, and phospholamban phosphorylation relieves the inhibition and increases calcium entry with a resulting increase in inotropy.21–24 In heart failure, phospholamban phosphorylation is decreased by an increase in unphosphorylated calcineurin,25 leading to decreased SERCA activity.26,27 A similar decrease in SERCA has been found in sepsis28 and in some forms of dilated cardiomyopathy.29 Cisternae store and release activator calcium, whereas longitudinal tubules remove calcium from the cytosol. Calcium release is primarily via the calcium-activated calcium release channel termed the ryanodine receptor. Both T-tubules and sarcoplasmic reticulum are sparse, undifferentiated, and disorganized early in gestation but increase and differentiate markedly late in gestation and after birth in mammals. Therefore the immature heart depends mainly on extracellular sources for activator calcium,15,19 partly explaining its marked calcium sensitivity.
Cytoplasm
During development, the proportion of mitochondria in the myocyte increases, particularly at the time of birth, and mitochondria become larger and develop more complex cristae.19 In the adult, approximately 30% to 40% of the muscle mass is made up of mitochondria. The cytosol contains other calcium-binding proteins19,30 and other major proteins such as tubulin and desmin.
Cytoskeleton and Extracellular Matrix
For contractile proteins to shorten the whole myocyte, they must be linked to both the cell membrane and the extracellular matrix. Longitudinal connections are made via the Z lines, representing discs that contain proteins such as α-actinin and filamin which connect the actin and titin filaments of adjacent myocytes.31,32 More lateral connections are made by the extrasarcomeric skeleton. There is an intermyofibrillar cytoskeleton with intermediate filaments, microfilaments, and microtubules.31,33–35 Desmin intermediate filaments provide a three-dimensional scaffold throughout the extrasarcomeric cytoskeleton and connect longitudinally to adjacent Z discs and laterally to subsarcolemmal costameres.33,35 Costameres are subsarcolemmal domains located in a periodic pattern, flanking the Z lines and overlying the I bands on the cytoplasmic side of the sarcolemma.36–40 They contain the focal adhesion-type complex, the spectrin-based complex, and the dystrophin/dystrophin-associated protein complex. The focal adhesion-type complex, made up of cytoplasmic proteins such as vinculin, ankyrin, and talin, connects with cytoskeletal actin filaments with transmembrane proteins such as the dystroglycans and the sarcoglycans.41–43 Dystrophin is linked to dystroglycan, laminin, and actin. These proteins help to fix sarcomeres to the lateral sarcolemma, stabilize the T-tubular system, and connect the sarcolemma to the extracellular matrix. Voltage-gated sodium channels colocalize with dystrophin, spectrin, ankyrin, and syntrophins. Potassium channels interact with the Z line and intercalated discs. In many of the genetic dilated cardiomyopathies, these proteins are abnormal,44–46 thereby explaining the abnormal muscle function.
Extracellular collagen plays a major role in cell-cell and cell-vessel interactions and in ventricular stiffness.47–50 With maturation, more collagen is type III and less is type I.51 The relationship between sarcomeres and cytoskeleton changes with maturation, perhaps accounting for maturational differences in the resting sarcomere’s mean length in myocytes.52 In addition, cell adhesion proteins stimulated by growth factors from the myocyte are in greatest amount in the neonate, decrease with postnatal age, but increase again during hypertrophy.53,54 Other elements in the extracellular matrix (e.g., laminin, fibronectin, and tenascin) play a major role during morphogenesis and during contraction55 and are important mediators in hypertrophy.
Nerves and Receptors
Adrenergic, muscarinic, and other receptors appear early and are functional even before innervation. Parasympathetic innervation precedes sympathetic innervation in all species.19,56,57 Innervation is present in the earliest viable human premature infants but may not be fully mature. Innervation is most advanced in species that are most independent immediately after birth.
Cardiac sympathetic nerve fibers come from cervical sympathetic and stellate ganglia. Right sympathetic nerves innervate the right and anterior surfaces of the heart. Left sympathetic nerves innervate the left and posterior surfaces. Vagal nerve fibers descending from medullary centers supply both atria and ventricles and the proximal portion of the bundle of His; the distal part of the bundle of His has only sympathetic nerve supply. Sympathetic and vagal afferents leave the heart and carry information from baroreceptors that respond to high pressures in the ventricles and to lower pressures in the atria, cavae, and pulmonary veins and from chemoreceptors that respond to locally produced substances such as bradykinin and prostaglandin.58
Ductus Arteriosus
The ductus arteriosus forms from the embryonic left sixth aortic arch and joins the main pulmonary artery that separates from the truncus arteriosus. The ductus is kept open by a balance between prostaglandin E2 (PGE2) and endothelin-1 (ET-1), both of which are formed in its wall and circulate from other sites. Initially, the ductus is very sensitive to the dilating action of PGE2, but later in gestation it becomes less sensitive to dilator and more sensitive to constrictor prostaglandins.59–61 After birth, oxygen reacts with a cytochrome P-450 and causes release of ET-1 (the most powerful ductus constrictor).62 A switch from dilator to constrictor prostaglandins occurs. In addition, oxygen modulates the function of mitochondrial electron chain transport by increasing the generation of H2O2, inhibiting voltage-gated potassium channels in ductus smooth muscle, thereby opening voltage-gated L-type Ca2+ channels to cause influx of calcium and ductus constriction.63–65 These constrictor effects overpower the dilating effect of nitric oxide, which is released from the ductus when oxygen tension rises.66 The ductus constricts, usually within the first 24 hours and almost invariably within 3 weeks. The lumen then becomes permanently occluded by fibrosis.61,67
Physiologic Development and Function
Myocardial Mechanics—Cardiac Sarcomere Function
Excitation-Contraction Coupling
When an electrical impulse reaches cardiac muscle, myocyte membranes depolarize. Extracellular calcium in high concentration at the sarcolemmal membrane and the T-tubules enters the cell rapidly. Spread of electrical excitation into the myocyte via the T-tubules also causes release of intracellular calcium from the sarcoplasmic reticulum.11,68,69
Cytosolic calcium increases from a concentration of 10−7 M in diastole to 10−5 M in systole. When the calcium that entered the cytosol binds to troponin C, the inhibitory effect of troponin I is antagonized, and a conformational change of troponin and tropomyosin exposes the actin-myosin binding sites.11,15,68,69 These sites interact with the myosin heads to form the cross-bridges (Figure 19-3). The myosin heads rotate, generate force, and move the actin filaments, just as oars move a boat through the water. Interaction between actin and myosin pulls the two Z lines toward each other, shortening the muscle and generating force. Increasing intracellular calcium results in greater cross-bridge formation and a greater generated force. Isoforms of the troponins and tropomyosin change during development, but the functional effects of these changes are unknown.19,70 However, troponin I is less sensitive to a fall in pH in the fetus than in the adult, which could be protective in perinatal acidosis.
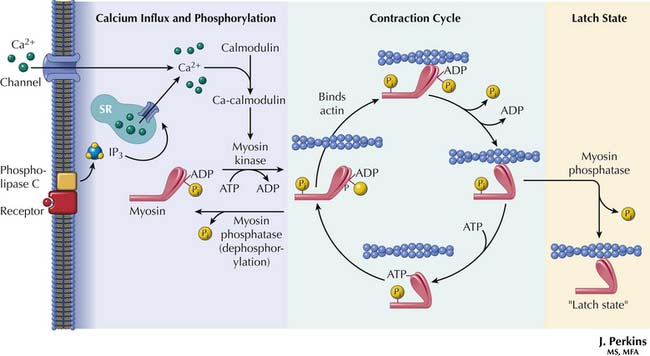
The myosin head contains an ATPase that liberates energy from ATP. The activity of the ATPase determines the velocity of shortening of unloaded muscle by affecting the rate of attachment and detachment of the cross-bridges.19,71 In most mammalian species, fetal myocardium contains V3 myosin isoform (having two β heavy chains) with a high ATPase activity rate. In humans and most of the larger mammals, however, almost all ventricular myocardial myosin is V3 isoform at any age, although human atria contain V1 myosin isoform.19
Sarcomere Length-Tension Relationships
Sarcomere length-tension relationships have been investigated in isolated cardiac muscle strips, usually papillary muscle with its nearly parallel fibers. The muscle strip is placed in a water bath. One end is tied to a lever and the other to a force transducer (Figure 19-4, A). Weights attached to the other end of the lever extend the muscle to any desired length before contraction (preload); excessive stretching is prevented by a stop. Other weights added to the lever after initial length is set affect the muscle only after contraction has started and so are termed the afterload. The muscle can be stimulated to contract by an electrical impulse. Instruments for measuring muscle length, sarcomere length by laser diffraction, calcium entry by various fluorescence methods, and a host of other specialized functions can be added.69,72,73
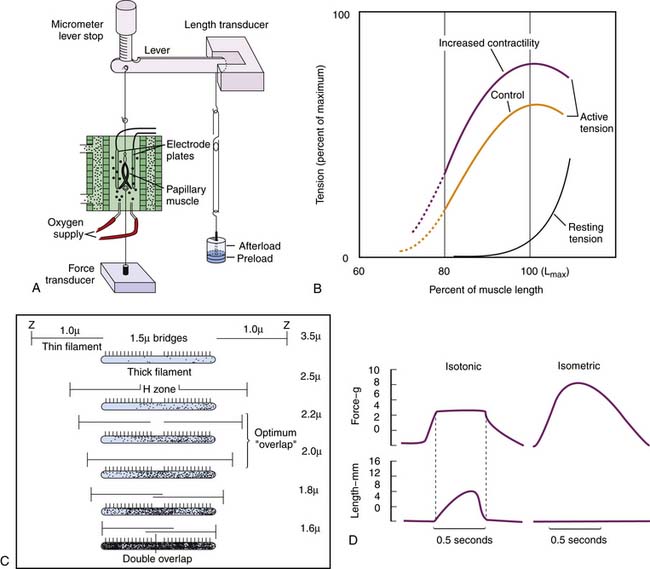
Stretching relaxed muscle produces an exponential-like increase in passive tension (Figure 19-4, B). This elasticity results mainly from titin.17,73–75 At very low sarcomere lengths, the actin filaments from each Z line overlap each other. As the sarcomere lengthens, the Z lines move farther apart and a gap appears between the two sets of actin filaments. When the sarcomere reaches a length of approximately 2.2 μm, there is a maximal overlap between actin and myosin filaments.13,69,71 (Figure 19-4, C). At longer muscle and sarcomere lengths, actin and myosin filaments overlap less. The maximal sarcomere length is 3.0 μm. Further elongation of the muscle occurs by slippage of fibers and not by further sarcomere lengthening.13,69,71
Active contraction is studied in two ways72 (Figure 19-4, D). First, muscle is stimulated to contract at different initial muscle lengths but is not allowed to shorten (isometric contraction). At the shortest lengths no force is generated; the muscle remains slack. As sarcomere lengths increase, force is generated and increases to reach a maximum at sarcomere lengths of approximately 2.2 μm. At longer sarcomere lengths, there may even be a decrease in force.71
If, at any length, passive tension is subtracted from the tension generated during isometric contraction, the resulting curve demonstrates active tension as a function of length (Figure 19-4, B).
Second, if afterload is small, the contracting muscle generates an appropriate force and then shortens while force remains constant (isotonic contraction). The rate of shortening is fastest at the onset of shortening, and from it, the velocity of shortening is measured (Figure 19-4, D). The shortening velocity ranges from zero when the load is so heavy that it prevents shortening to a maximum when the external load is zero69; however, true zero loading is impossible because of internal viscosity and elastic forces.19,71 Increases in cytosolic calcium increase the force generated during contraction but have little influence on the maximal velocity of shortening.
In fetal lambs, the passive tension of muscle strips is abnormally high. Active tension per mm2 cross-sectional myocyte area at any given afterload is below adult values but is proportional to the reduced number of myofibrils.19,30,57 Contractile material accounts for approximately 60% of cardiac muscle in adults, but only 30% in fetuses. The extent and velocity of shortening are reduced in fetal heart muscle, but correcting for the amount of contractile machinery suggests the intrinsic performance of fetal and adult actin-myosin filaments is similar.19 The change from fetal to adult performance seems to occur fairly soon after birth, when the myofibrillar array becomes regular and when the T-tubules and the sarcoplasmic reticulum develop into their adult form.19 For this reason, prematurely born infants (and, to a lesser extent, full-term infants) have a much-reduced ability to tolerate an increase in afterload and are exquisitely sensitive to reductions in serum calcium concentrations.
Myocardial Mechanics—Myocardial Receptors and Responses to Drugs
α1-adrenoceptors appear early in gestation and in many species reach their highest density in the neonate.19,56,76 These developmental changes may be associated with the normal cell hypertrophy that occurs during development. By contrast, β-adrenoreceptors increase progressively with age. Both β1 and β2 are present on myocytes.77,78 In addition, histamine H2, vasoactive intestinal polypeptide (VIP), adenosine A1, acetylcholine M2, and somatostatin receptors have been identified. They act on the myocyte’s contractile apparatus through one of two main pathways.
The major pathway involves the membrane-bound receptor–G protein–adenylate cyclase complexes. G proteins include the Gs (stimulatory) and Gi (inhibitory) proteins.79 In their inactive state, these G proteins include α, β, and γ subunits and guanine diphosphate (GDP). When agonists stimulate β-adrenergic, histamine, or VIP receptors, the G proteins undergo a conformational change. The changes induce the Gs protein to exchange its GDP for guanine triphosphate (GTP) and release the β and γ subunits. The Gs-α-GTP complex interacts with adenylate cyclase to convert ATP to cyclic adenine monophosphate (cAMP), which activates a variety of protein kinases to phosphorylate proteins including voltage-dependent calcium channels, phospholambin, and troponin I. Consequently, calcium entry during depolarization and during uptake of calcium into the sarcoplasmic reticulum storage pool is increased, thus increasing contractility. The Gs-α-GTP complex has intrinsic GTPase activity that converts GTP to GDP. The β and γ subunits rejoin the complex, which now is available for further activation by the receptor. In this way, as long as receptors are occupied by the agonist, the Gs cycle produces increasingly more cAMP, thereby amplifying the stimulatory signal. The Gi protein complex undergoes a similar cycle when adenosine, acetylcholine, or somatostatin receptors are stimulated; however, activating Gi protein reduces cAMP formation and decreases contractility. β2 adrenergic receptors also couple to Gi in addition to Gs.80 Gi in this context is thought to oppose the effects of Gs to some degree, including limitation of the acute positive inotropic response to adrenergic stimulation and offering some protection from apoptosis.81–83
Another signal-transducing system in the human heart is the phospholipase C-diacylglycerol-inositol triphosphate pathway, activated by α1-adrenergic and M2-muscarinic receptors.84–86 Occupation of the receptors activates phospholipase C, which cleaves phosphatidylinositol triphosphate in the cell membrane to produce diacylglycerol and inositol triphosphate. The former activates protein C kinase in the membrane, which may hinder the effects of cAMP. The latter facilitates calcium release from the sarcoplasmic reticulum. This pathway is important in smooth muscle contraction but is of less importance in heart muscle.
In heart failure, the number of β1-adrenergic and VIP receptors are down-regulated, and β2-adrenergic receptors are uncoupled from G proteins.21,77,78,87,88 These changes make the myocardium less responsive to circulating or locally released catecholamines or VIP and play a role in the reduced contractility observed in heart failure. Treating heart failure with β-adrenergic blocking agents not only has reversed the receptor changes, but has also been associated with improved function of muscle strips in adult patients.89–92
Coupling of β-adrenoreceptors is incomplete at birth. Milrinone is an agent that stimulates contractility by inhibiting phosphodiesterases; it bypasses the adenylate cyclase system. Although previously thought to be ineffective in the newborn,93 a multicenter randomized trial94 and subsequent widespread use has confirmed its efficacy in the neonatal population.95,96 Since contractile mechanisms are almost fully developed at birth, the majority of mechanisms controlling contractility (except for changes in the source of calcium) are in place at birth.
Myocardial Mechanics—Integrated Muscle Function
Relationship Between Muscle Strips and Intact Ventricles
Preload stretching a muscle strip is equivalent to end-diastolic fiber length of the intact ventricle. This length can be measured by various devices in animals, but in the intact human ventricle it is best related to end-diastolic diameter or volume. Frequently, end-diastolic pressure has been used interchangeably with end-diastolic volume as an index of preload, but this usage can be misleading if the distensibility of the ventricle changes or if pressure outside the heart (pericardial or intrathoracic) rises.97–100
Afterload is more complicated in the intact ventricle. Commonly, aortic systolic pressure is equated with afterload. However, in the muscle strip, afterload represents the force exerted by the muscle during contraction, and pressure and force are not the same.75,101,102 It is preferable to calculate circumferential wall stress, which at the midwall is a function of ventricular pressure, diameter, and wall thickness. Both peak systolic and end-systolic wall stress can be used to assess ventricular function.
Calculations of wall stress are based on the Laplace relationship:

where P is pressure, r is radius of curvature, and h is wall thickness. Because the left ventricle is not a regular sphere, particularly in systole, the Laplace formula is an oversimplification.103 A fairly simple and accurate formula was developed by Grossman and colleagues104:
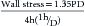
where P is pressure, D is left ventricular minor axis dimension, and h is wall thickness at the level of the minor axis. This equation can be written as:
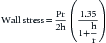
that is, as the Laplace equation modified by the expression in parentheses. Note that if the left ventricle dilates acutely, wall stress rises markedly because r gets bigger and h gets smaller.
The major findings from studies of muscle strips have been confirmed in intact ventricles. Increasing preload increases the pressure generated by an isolated ventricle that is not allowed to eject, as observed in the last century by Otto Frank. If the ventricle is allowed to eject, then increased preload allows the heart to eject the same stroke volume against an increased afterload or else to eject a greater stroke volume against a constant afterload. This is the Starling component of the Frank-Starling law.105,106 The mechanism of this response is twofold: (1) lengthening the sarcomere narrows it and places the myosin and actin fibrils closer together for stronger interaction, and (2) increased calcium sensitivity is mediated in some way by titin stretching.18 If an inotropic drug is given, then contractility increases and from a given fiber length greater force of contraction is achieved. This is a phenomenon seen every day in the intensive care unit.
The force-frequency relationship can be determined in intact hearts107,108 by examining the response of the maximal rate of change of pressure (dP/dt max) in the ventricles after premature beats. The results in intact ventricles and muscle strips are similar. Subsequently, Seed and colleagues109 applied this technique to humans with normal or abnormal left ventricular function and found an optimal R-R interval of 800 ms. They also examined dP/dt max for two beats given at optimum intervals after the premature stimulus. As expected, the first normal beat after the premature beat was potentiated because the extra calcium introduced into the cytosol by the premature beat was available to potentiate the first beat after the premature stimulus. The second postpremature beat also was potentiated, but less so. They used the ratio of the potentiation of these two beats to calculate the fraction of calcium recirculating from one beat to the next. This amount was constant in any one patient but was much less in those with left ventricular dysfunction.
Pressure-Volume Loops
The modern approach to analyzing these loops is based on the elastance concept of Suga and Sagawa.110–112 Elastance is the ratio of pressure change to volume change. Consider an isolated ventricle containing a balloon that can be inflated to different volumes. At each volume the ventricle is stimulated to contract and generates a peak systolic pressure (Figure 19-5, A). As volumes increase, so do the peak systolic pressures generated, and the relationship is linear (Frank’s law). The line joining the peak pressures intercepts the volume axis at a positive value, termed V0, that indicates the unstressed volume of the ventricle. The equation for this line is as follows:
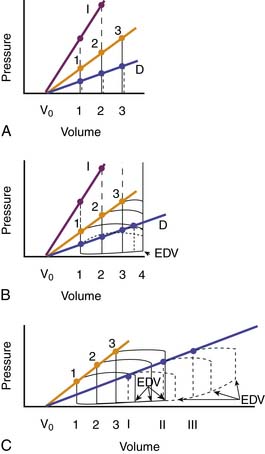
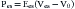
where Pes is end-systolic pressure, Ees is slope of the line, Ves is end-systolic volume, and V0 is unstressed volume. If contractility increases (more calcium enters the cells), the ventricle can generate greater pressures at any given volume, thereby generating a steeper pressure-volume line (higher value of Ees; dashed line I in Figure 19-5, A). If contractility decreases, the ventricle generates lower pressures at any given volume, and the pressure-volume line is less steep (lower value of Ees; dotted line D in Figure 19-5, A). Ees is also termed Emax.
If the ventricle is allowed to eject, the typical pressure-volume loop shown in Figure 19-5, B, is seen. During diastolic filling, volume increases and diastolic pressure rises slightly because of the increase in passive tension. At the end of diastole, isovolumic systole occurs, and ventricular pressure rises with no change in volume. When ventricular pressure exceeds aortic pressure, the aortic valve opens, blood is ejected, and ventricular volume decreases. Ejection ends, and pressure falls to diastolic levels as isovolumic relaxation occurs. The pressure and volume reached at the end of systole are those that would have been attained by the isolated ventricle at that same end-systolic volume. In other words, at a given volume, no higher pressure can be generated (loop 1; Figure 19-5, B). The decrease in volume during ejection is the stroke volume which, divided by the end-diastolic volume, gives the ejection fraction; normally, ejection fraction is greater than 65%.
If afterload is suddenly increased by raising aortic pressure, the normal heart responds as shown in Figure 19-5, B. In the first beat after the increase, the ventricle has to generate a higher pressure before the aortic valve opens (loop 2). It then ejects but cannot eject a normal stroke volume because that would require higher pressure from the same end-diastolic length (preload). In fact, the end-systolic volume is that which is appropriate for the higher pressure (compare Figure 19-5, A and B). If different afterloads are used, the end-systolic pressure-volume points define a sloping line that is the same as the line obtained in the isolated heart at those same volumes; this is the maximal ventricular elastance (Ees) or end-systolic elastance (Ees) line. If ventricular contractility increases, then the ventricle can attain higher ejection pressures at any given volume, and the end-systolic pressure-volume points lie on a steeper line that lies above and to the left of the normal line (purple line I in Figure 19-5, B). If ventricular contractility decreases, then the ventricle cannot generate normal pressures at any given end-diastolic volume, and the end-systolic pressure-volume line lies below and to the right of the normal line (blue line in Figure 19-5, B). Note from Figure 19-5, B, that, from a given end-diastolic volume, the ventricle with impaired contractility can either eject a normal stroke volume at much reduced pressures or eject at a normal pressure only by reducing its stroke volume drastically (loop 4).
In beats that follow a sudden increase in afterload, the ventricles adjust. Because of the reduced stroke volume in the first beat, the end-systolic volume is larger than normal. During diastole, however, a normal stroke volume enters the ventricle so that end-diastolic volume increases (loop 2 in Figure 19-5, C). In normal ventricles, the increased end-diastolic fiber length causes little increase in diastolic pressure. The pressures during ejection and the end-systolic pressure-volume point are unchanged, but stroke volume and ejection fraction increase. After a few more cycles, a new equilibrium is established (loop 3) in which the ventricle ejects a normal stroke volume at the higher afterload. The ejection fraction, however, is subnormal because although the stroke volume is normal, the end-diastolic volume is increased. The ventricle has adapted to the higher afterload by increasing end-diastolic fiber length, a phenomenon described by Starling and discussed by Ross101,102 under the term preload reserve. If the ventricle has decreased contractility (dashed loops), the same pattern of response occurs, but with some important differences. With decreased contractility, the ventricle cannot eject a normal stroke volume from a normal end-diastolic volume. Compensation results in a larger than normal increase in end-diastolic volume, even at normal afterloads. Any increase in afterload causes a further increase in end-diastolic volume, and this increase causes diastolic pressures to rise to high values that cause pulmonary congestion. The normal preload reserve has been used up in the attempt to eject a reasonable stroke volume against a modestly increased afterload. In more depressed hearts, even normal afterloads cannot be handled by the ventricle without a pathologically raised diastolic pressure in the ventricles or a drastic decrease in stroke volume. Note that in these hearts, because of the relatively flat slope of the maximal ventricular elastance line, a slight reduction of afterload produces a relatively large increase in stroke volume and a relatively large decrease in ventricular end-diastolic volume and pressure. This is one of the mechanisms for cardiac improvement with afterload reduction.
The normal right ventricular (RV) pressure-volume curve is triangular in shape, unlike the more rectangular left ventricular (LV) pressure-volume curve.113 This difference is accounted for by a relative lack of isovolumic contraction and relaxation times in the RV. The normally low afterload of the RV and the high compliance of the outflow portion of the ventricle allow ejection to begin almost instantaneously after the onset of contraction and proceed through pressure decline so that there is near complete emptying of the ventricle by the end of systole and the ejection time of the RV thus spans the entire period of systole. An important consequence of this relationship is that even small increases in RV afterload begin to make the RV pressure-volume curve begin to resemble the normal LV pressure-volume curve, with isovolumic contraction and relaxation times becoming more prominent.114 Ejection fraction is reduced, although stroke volume may be maintained due to RV dilation,115 and the thin-walled RV may handle this new physiology quite poorly.
Assessing Myocardial Contractility
An index of contractility must reflect the ability of the ventricle to perform work independent of changes in preload and afterload. Contractility can perhaps best be defined as the alterations in cardiac function that occur secondary to changes in cytosolic calcium availability or sarcomere sensitivity to calcium. Thus, β-adrenergic agonists or phosphodiesterase inhibitors, which increase cytosolic calcium, and thyroxine, which alters myosin ATPase sensitivity to calcium in some species by altering the dominant isoform, are positive inotropic agents. However, quantifying contractility in the intact heart or assessing contractile effects of an intervention is difficult72 because all indices of contractility are indices of overall performance and are not independent of the other determinants of performance. For example, cardiac output is an excellent index of the systolic performance of the intact ventricle, but it is not a useful index of contractility because of its high sensitivity to preload, afterload, and heart rate. It is convenient to divide methods of assessing contractility into those based on early events in the cardiac cycle (isovolumic phase indices) and those that occur later (ejection phase indices).
Isovolumic Phase Indices. The concept of maximal velocity of contraction against zero load (Vmax) once was popular, but the complexity of the mechanics of cardiac muscle made it difficult to assess what would have been the true index, namely, Vmax of the contractile element alone.116 In practice, too, it is not possible to abolish internal loading of the muscle fiber. Applying this concept to the intact heart was even more difficult.117
Ejection Phase Indices. The index of contractility most commonly used today is the maximal (end-systolic) ventricular elastance of Suga and Sagawa, which is independent of changes in preload (see previous text). Several different afterloads must be obtained, and either ventricular volumes must be measured or echocardiographic dimensions must be used as substitutes for volumes. The most clear-cut results have been obtained when reflex changes in contractility are prevented, which may explain why the relationship is less well established in conscious than in anesthetized animals.75,118 Several studies have shown that the maximal elastance line often is alinear and gives a negative intercept on the pressure axis, that is, a negative resting volume.119,120 To deal with this simply, some investigators use the values of Emax in the mid-range of pressures.120
Ejection fraction and velocity of shortening also provide information about ventricular function. Because these two variables depend on afterload (see Figure 19-5), it is necessary to adjust for changes in afterload. This adjustment has been made in adults and children121–126 by providing normal data for the relationship between end-systolic wall stress and either velocity of shortening or ejection fraction (Figure 19-6). However, the relationship is not linear,127 so single-point determinations are of little use.
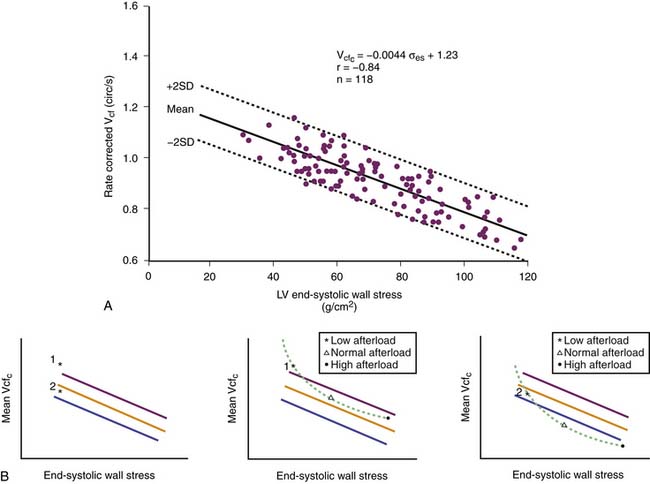
Ventricular Function Curves
Sarnoff and Mitchell128 introduced the ventricular function curve. They measured LV diastolic pressure and stroke work, then infused fluids and examined the relationship between the two variables (Figure 19-7, A). If contractility increased, the curve shifted up and to the left; at any end-diastolic pressure, a greater stroke work was achieved. If contractility decreased, the curve shifted down and to the right. One problem with this technique, recognized by Sarnoff, was that curvilinearity of the diastolic length-pressure relationship produced an S-shaped curve when relating stroke work to end-diastolic pressure and that techniques for measuring fiber length or ventricular volume were inadequate. In addition, using pressure instead of fiber length or volume may lead to misinterpretations if pericardial or pleural pressures change substantially. Several groups of investigators adapted this function curve to examine the stroke volume to end-diastolic pressure relationship, but this is even less satisfactory because stroke volume is affected by the resulting increases of afterload.
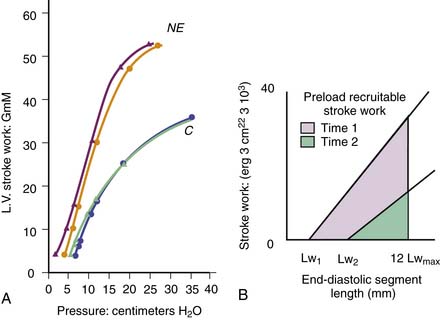
Figure 19–7 Ventricular function curves. A, Typical function curve relating left ventricular diastolic pressure to left ventricular (external) stroke work. C, Control state; NE, increased contractility resulting from norepinephrine infusion. Pairs of lines indicate repeatability of the measurement. (Redrawn from Sarnoff SJ, Mitchell JH: The control of the function of the heart. In Hamilton WF, Dow P, editors: Handbook of physiology, section 2: circulation, vol 1, Washington, DC, American Physiological Society, 1962.) B, Preload recruitable stroke work area, in which the area under the curve relating end-diastolic segment length to stroke work is indicated for two different contractile states of the ventricle. Lw1, Lw2, Intercepts on the x axis; Lwmax, maximal value of Lw for the whole experiment. For details, see reference.
(Adapted from Glower DD, et al: Am J Physiol 255:H85, 1988.)
More recently, the relationship of stroke work to end-diastolic fiber length or ventricular volume has been examined in conscious dogs with autonomic blockade.105 This relationship, termed preload recruitable stroke work, was linear and independent of changes in afterload (Figure 19-7, B). The line intercepted the length or volume axis at values close to the unstressed length or volume, that is, at the length or volume that the ventricle has at zero transmural pressure. Calcium infusion increased the slope of the line without changing the intercept on the length axis. Subsequently, the same group extended this analysis to ischemic ventricles.129 Depression of ventricular function shifted the stroke work to end-diastolic segment length relationship to the right (increased intercept) and decreased the slope. This concept and the ventricular elastance concept have much in common. Both require measurements of wall force, stroke work, ventricular volume, or minor axis diameter and changing them over a range so that the lines or areas defining these indexes can be obtained.
Pericardial Function
The parietal pericardium is a stiff membrane that surrounds the heart loosely, separated from it by a small amount of lubricating pericardial fluid. Intrapericardial pressure is negative, reflecting the negative intrapleural pressure. On the other hand, the pericardium exerts a surface pressure on the heart that would exist even if all fluid were removed. If the pericardium had holes in it, fluid would leak out and there would be no fluid pressure, but the heart still could be compressed. This surface pressure varies in different regions but, in general, is similar to right atrial pressure.99,130 As a result, in the usual situation transmural diastolic pressure across the wall of the left ventricle is not the same as left ventricular diastolic ventricular pressure. It can be estimated by subtracting right atrial pressure from LV pressure. The pericardium can restrict dilatation of the LV if there is a tense pericardial effusion (tamponade)131 and if the ventricles dilate acutely. Thus if the ventricles enlarge because of sudden volume load or sudden myocardial depression, the pericardium becomes tense and restrains further enlargement of the ventricles.99,100
In some patients with acute myocardial ischemia, left ventricular diastolic pressure can be greatly increased without much change in ventricular volume because of tension in the pericardium. This mechanism makes it difficult to interpret changes in diastolic pressure-volume relations only in terms of myocardial stiffness.97,98,130,132,133






Full access? Get Clinical Tree


