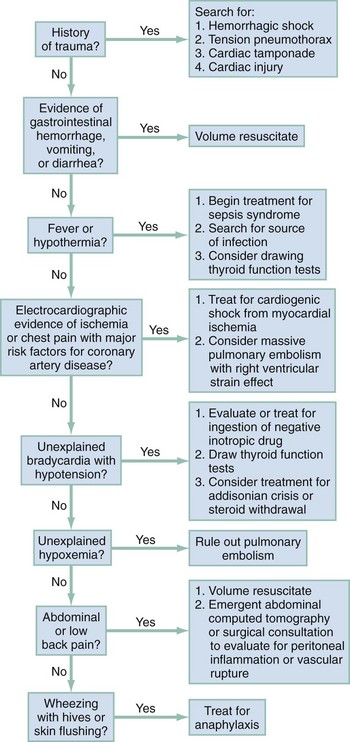
Chapter 6 In philosophic terms, shock can be viewed as a transition between life and death. Whether shock results from hemorrhage, sepsis, or cardiac failure, mortality rates exceed 20%.1,2 In scientific lexicon, shock results from the widespread failure of the circulatory system to oxygenate and nourish the body adequately. In the laboratory the scientist defines the metabolic effect of shock quantitatively, by examining the mechanisms by which shock alters mitochondrial energy transfer, evokes the production of toxic chemicals, and reduces their removal. At the bedside, however, the clinician identifies shock by linking the clinical impression, synthesized from the patient’s history of present illness, age, underlying health status, and general appearance, to quantitative data, including vital signs, blood chemistry, urine output, and direct measurements of oxygenation. When the clinical impression and the quantitative data suggest widespread organ hypoperfusion, emergent resuscitation is used to restore normal tissue oxygenation and substrate delivery to prevent deterioration into systemic inflammation, organ dysfunction, and death. At the subcellular level, shock first affects the mitochondria. Mitochondria function at the lowest oxygen tension in the body, but paradoxically, they consume almost all the oxygen used by the body. More than 95% of aerobic chemical energy comes from mitochondrial combustion of fuel substrates (fats, carbohydrates, ketones) plus oxygen into carbon dioxide (CO2) and water. Mitochondria therefore have been referred to as the “canaries in the coal mine” because they are affected first in conditions of inadequate tissue perfusion.3,4 When mitochondria have inadequate oxygen, the cell catabolizes fuels to lactate, which inexorably accumulates and diffuses into the blood. For years, shock has been classified into four broad categories based on Blalock’s 1934 description: hematologic, neurologic, vasogenic, and cardiogenic. This basic organization scheme remains useful today. Box 6-1 outlines five categories of shock that generally have specific mechanisms and treatments. The epidemiology of shock in the emergency department context remains speculative because shock is rarely listed as a primary coding diagnosis and depends on defining criteria. Arterial hypotension, defined as a systolic blood pressure (BP) below 100 mm Hg, is measured at least one time in 19% of ED patients5; however, diagnosed traumatic, cardiogenic, or septic shock is less common, constituting about 1 to 3% of all ED visits. The second phase of organ injury from hemorrhagic shock occurs during resuscitation. It has been said that the acute phase of hemorrhage “cocks the gun” by initiating the inflammatory cascade, and resuscitation “pulls the trigger” by accentuating the inflammation-induced organ injury from hemorrhagic shock. During resuscitation, neutrophils become most aggressive, binding to the lung endothelium and causing capillary leaks that characterize acute respiratory distress syndrome (ARDS). Inflammatory cytokines are liberated during resuscitation, and membrane injury occurs in many cells. In the liver, damage from inflammation and reactive oxygen species from neutrophils is compounded by persistent microischemia. During resuscitation from hemorrhagic shock, the normal balance of vasodilation by nitric oxide (NO) versus vasoconstriction by endothelins becomes distorted, producing patchy centrilobular ischemic damage in the liver, which may produce an immediate rise in blood transaminase levels. A growing body of evidence suggests that resuscitation from hemorrhage exerts greater injury on the heart than the actual hypotensive insult.6 Depending on the degree of hypotensive insult, the kidney may manifest acute spasm of the preglomerular arterioles, causing acute tubular necrosis. Systemic metabolic changes can impair fuel delivery to the heart and brain, secondary to depressed hepatic glucose output, impaired hepatic ketone production, and inhibited peripheral lipolysis. 1. More patients are being treated at home for chronic immunocompromising diseases with indwelling catheters, which can serve as portals of entry into the vascular space for Staphylococcus aureus and coagulase-negative staphylococci. 2. The frequency of community-acquired infections caused by antibiotic-resistant gram-positive organisms has greatly increased in recent years, including infections caused by S. aureus, Streptococcus pneumoniae, and Streptococcus pyogenes. Septic shock often causes three major effects that must be addressed during resuscitation: relative hypovolemia, cardiovascular depression, and induction of systemic inflammation. Septic shock produces relative hypovolemia from increased venous capacitance, which reduces right ventricular filling. Septic shock often causes absolute hypovolemia from gastrointestinal volume losses, tachypnea, sweating, and decreased ability to drink during development of the illness. Sepsis also induces capillary leak, which leads to relative loss of intravascular volume into third spaces. Recent evidence has shown that septic shock causes myocardial depression simultaneously with vasodepression and capillary leak. Direct measurements of cardiac contractility have shown that cardiac mechanical function becomes impaired early in the course of septic shock, even in the hyperdynamic stages. Multiple mechanisms may explain depressed heart function in sepsis, including actions of specific cytokines (most notably tumor necrosis factor alpha [TNF-α] and interleukin 1 beta [IL-1β]), overproduction of NO by nitric oxide synthase (iNOS),7 and possibly impairment in mitochondrial oxidative phosphorylation coincident with reduced mechanical efficiency.8,9 Evidence indicates that circulating mediators, myocardial cellular injury from inflammation, and deranged metabolism interact synergistically to injure the heart during septic shock. Systemic inflammation causes capillary leak in the lung, which may cause alveolar infiltration characteristic of ARDS early in the treatment of septic shock in up to 40% of patients. With the potential for early development of ARDS, more profound ventilation-perfusion ( Cardiogenic shock results when more than 40% of the myocardium undergoes necrosis from ischemia, inflammation, toxins, or immune destruction. Otherwise, cardiogenic shock essentially produces the same circulatory and metabolic alterations as are observed with hemorrhagic shock. Undoubtedly, impaired baseline cardiac function can contribute to the development of circulatory shock secondary to infection, hemorrhage, or vasodilatory drug overdose. However, when shock results from a pure cardiac cause, severe left ventricular dysfunction will be evident on echocardiography early in the course. Patients with severe dysfunction are far more likely to have a cardiogenic cause of shock than patients with normal or moderate left ventricular dysfunction.10 Patients in the ED frequently are in shock with no obvious cause. Rapid recognition of shock requires the integration of information from immediate history and physical examination, and shock can be strongly supported by the presence of a worsening base deficit or lactic acidosis. In general, patients with shock exhibit a stress response: they are ill appearing, asthenic, pale, often sweating, and usually tachypneic or grunting, and often have a weak and rapid pulse (Box 6-2). HR can be normal or low in shock, especially in cases complicated by prescribed drugs that depress HR or by profound hypoxemia. BP initially can be normal because of adrenergic reflexes. Although arterial BP as a sole measurement remains an unreliable marker of circulatory status, the finding of a single systolic BP less than 100 mm Hg in the ED is associated with a threefold increase in in-hospital mortality and a tenfold increase in sudden and unexpected death.5 The HR/systolic BP ratio may provide a better marker of shock than either measurement alone; a normal ratio is less than 0.8. Urine output provides an excellent indicator of organ perfusion and is readily available with insertion of a Foley catheter into the bladder. Measurement of urine output, however, requires 30 minutes to 1 hour for accurate determination of whether output is normal (>1.0 mL/kg/hr), reduced (0.5-1.0 mL/kg/hr), or severely reduced (<0.5 mL/kg/hr). Point measurements of the arterial or venous lactate concentration and the base deficit can be rapidly performed and provide accurate assessment of global perfusion status. A lactate concentration greater than 4.0 mM or a base deficit more negative than −4 mEq/L predicts the presence of circulatory insufficiency severe enough to cause subsequent multiple organ failure.11,12 Once the empirical criteria for circulatory shock have been discovered, the next step is to consider the cause of the shock. Figure 6-1 shows a potential sequence of decisions to help arrive at a diagnosis in a patient with undifferentiated shock. The history, vital signs, and physical examination documented by prehospital providers afford valuable insight into a patient’s physiologic status before any medical intervention and can be useful in ED management. Studies suggest that both medical and trauma patients with prehospital hypotension have a threefold to fourfold higher in-hospital mortality rate than patients without hypotension.13,14 Laboratory, radiographic, and other ancillary data should be ordered to assess tissue and vital organ perfusion and to diagnose injury from trauma, find the source of infection with sepsis, or identify the cause of cardiac failure. A chest radiograph, electrocardiogram, finger-stick glucose measurement, complete blood count (CBC), urinalysis, serum electrolytes, and kidney and liver function tests are all indicated in the ED assessment. Arterial blood gases are ordered for a base deficit calculation and to correlate arterial gas tensions (oxygen [PaO2] and PaCO2) with those measured by pulse oximetry and capnography. Serum lactate measurement should be performed as early as possible in patients with suspected shock. Either venous or arterial lactate concentrations can be used.15,16 If peripheral venous lactate is used, the effect of time, storage temperature, and tourniquet use have no significant effect on in vitro lactate production by erythrocytes if the measurement is done within 15 minutes after the sample has been obtained.17 Cardiac and abdominal bedside ultrasound scanning can screen for inadequate central venous volume, occult hemoperitoneum, abdominal aortic aneurysm, left ventricular failure, and cardiac tamponade.18 A systematic ultrasound protocol can significantly improve the physician’s ability to accurately diagnose the cause of undifferentiated shock in ED patients, and the finding of hyperdynamic left ventricular function in patients with undifferentiated shock strongly suggests sepsis as the cause.19,20 Consensus definitions of shock show the spectrum of hypoperfusion for the following three common causes of shock (Box 6-3): BOX 6-3 Definitions and Criteria for Septic, Hemorrhagic, and Cardiogenic Shock Systemic Inflammatory Response Syndrome (SIRS) 1. Temperature >38° C or <36° C 3. Respiratory rate >20 breaths/min or PaCO2 <32 mm Hg 4. White blood cell count >12,000/mm3, <4000/mm3, or >10% band neutrophilia
Shock
Perspective
Classification
Epidemiology
Specific Causes
Septic Shock
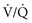 ) mismatching, and pneumonia or pulmonary aspiration, hypoxemia is more severe with septic shock than hemorrhagic shock.
) mismatching, and pneumonia or pulmonary aspiration, hypoxemia is more severe with septic shock than hemorrhagic shock.
Cardiogenic Shock
Clinical Features

Full access? Get Clinical Tree