Shock: Introduction
Shock is a state of severe systemic reduction in tissue perfusion characterized by decreased cellular oxygen delivery and utilization as well as decreased removal of waste byproducts of metabolism. Hypotension, although common in shock, is not synonymous to shock. One can have hypotension and normal perfusion, or shock without hypotension in a patient who is usually very hypertensive. Shock is the final preterminal event in many diseases. Progressive tissue hypoxia results in loss of cellular membrane integrity, a reversion to a catabolic state of anaerobic metabolism, and a loss of energy-dependent ion pumps and chemical and electrical gradients. Mitochondrial energy production begins to fail. Multiple organ dysfunction follows localized cellular death, and organism death follows. Despite recent advances in treatment, mortality remains high: > 50% in cardiogenic shock and > 35% in septic shock.
Pathophysiology of Shock
Blood pressure is determined by the formula BP = systemic vascular resistance (SVR) × cardiac output (CO), where CO = heart rate (HR) × stroke volume (SV). SV = end diastolic volume (EDV) minus end systolic volume (ESV). EDV is the filled ventricular volume prior to systolic contraction averaging about 100 cc in many adults. ESV is residual blood left in the ventricle after emptying during systole averaging about 40 cc. Therefore, the determinants of blood pressure are vascular resistance, HR, preload volume, and contractility (see Figure 11–1). SVR is the vascular “tone” and is a large determinant of diastolic blood pressure. EDV is largely determined by preload volume that augments SV via Frank–Starling curves where increases in diastolic filling volumes increase CO. ESV is determined largely by cardiac contractility and it decreases as the heart ejects a greater percentage of its diastolic volume. For example, one can increase SV by increasing preload (EDV) with volume or decreasing ESV with increased contractility. The ejection fraction ((EDV – ESV)/EDV) thus increases.
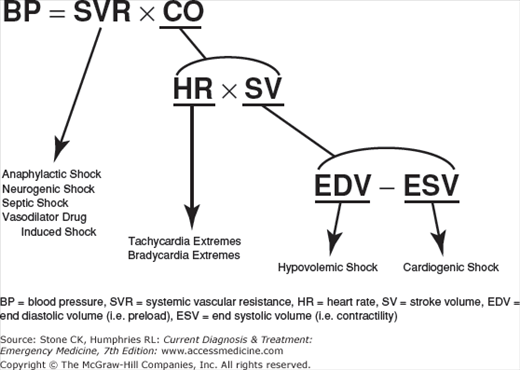
The initial derangement precipitating a state of shock might be (1) vasodilation (causing a decreased SVR) from sepsis, anaphylaxis, drugs, or cervical cord lesion, (2) extremes of HR, (3) loss of preload volume (causing decreased EDV) from blood or volume loss, or (4) loss of contractility (increasing the ESV) from heart failure. Compensatory mechanisms come into play and provide many of the clinical clues to early shock.
The initial compensatory mechanisms depend on the initial insult. (1) Vasodilation with loss of SVR generally causes a compensatory tachycardia and thirst. Despite systemic tissue hypoxemia, the skin remains perfused and is warm initially. (2) Blood or fluid loss (decreasing EDV) causes a reflex increase in SVR, which increases diastolic BP, narrowing the pulse pressure, increases sympathetic cholinergic sweating and makes the patient pale, thirsty, and cool. As volume loss increases, tachycardia and hypotension ensue. (3) Loss of contractility also is compensated by increases in SVR to maintain blood pressure with similar symptoms.
Once compensatory mechanisms fail, irreversible shock occurs with irreversible cell death, microcirculation plugging, and free radical generation. There is loss of autonomic regulation due to local nitric oxide vasodilator generation, and even with complete correction of blood volume (for example, in hypovolemic shock), tissue function, and organ function are not restored, causing eventual death.
Causes of Shock
The major classical classification of shock includes (1) hypovolemic, (2) cardiogenic, (3) distributive, and (4) obstructive shock. The first three involve a primary derangement in EDV, ESV, and SVR, respectively, while obstructive shock is usually a problem with SV due to mechanical obstruction to preload. Causes of “obstructive shock” are usually classified as hypovolemic or cardiogenic. Common causes of each type are listed in Table 11–1.
|
Clinical Presentation
There is no one clinical or biological test to determine shock. If compensatory mechanisms are functioning early in shock, one may not see hypotension but instead an anxious patient still maintaining a blood pressure. In these early stages (called preshock) symptoms can be subtle, but provide an opportunity for early intervention. Waiting for full-blown shock leads to a loss of precious time, and an aggressive proactive approach should be pursued.
During the early or preshock state, pale, cool, moist skin reflects compensatory elevated SVR in hypovolemic and cardiogenic shock. The pulse pressure narrows (with a slight decrease in systolic blood pressure and rise in diastolic blood pressure) and patient anxiety increases. Blood is shunted preferentially from “nonessential” skin and gastrointestinal (GI) tract to heart and brain. After a 20–30% volume loss in hypovolemic shock, tachycardia increases, urine volumes decrease with decreased renal blood flow, and the patient becomes more agitated. In cardiogenic shock, left-sided heart failure manifests as pulmonary edema and right-sided heart failure manifests as peripheral edema with elevated jugular venous distention (JVD).
In distributive shock, the primary problem is loss of vascular tone with erythematous warm skin despite hypotension. Tachycardic response is variable and early on, the heart may be hyperdynamic. This is the “warm patient in shock.”
In full-blown shock, patients become agitated and finally decrease their mental status. Hypotension occurs and may be profound. The patient has tachypnea until respiratory failure occurs and has a metabolic acidosis due to elevated lactic acid from anaerobic metabolism. At the cellular level, tissue oxygen extraction is maximal and is reflected in decreased mixed venous O2 saturation. Multiple organ failure follows. Irreversible shock follows if treatment is not aggressive.
Initial Evaluation of Shock
In the initial evaluation of a patient in preshock, one must ask: Is the patient in shock or headed that way? It is ill advised to wait for severe hypotension in the patient who is still compensating for their shock before aggressively intervening. The clinician’s first priority is to maintain vital functions while exploring the potential causes of shock. One should also consider early decontamination if the patient has been exposed to a toxin.
ABCs open the airway and maintain adequate ventilation/oxygenation with high flow oxygen. Airway adjuncts such as the nasopharyngeal airway may help, but one must anticipate worsening of the airway with time. If the patient cannot protect the airway, has a GCS score < 9 in trauma, has extremes of respiratory rate or is hypoxic despite supplemental oxygen, endotracheal intubation is indicated. Relieve any tension pneumothorax based on clinical grounds (below). Establish multiple short-length, large-bore peripheral IV access and place on a cardiac monitor in a critical care area of the ED. Central venous access and arterial catheter placement should be considered, but should not delay resuscitation. Intravenous access in shock can be challenging and intraosseous access may be timely. Remove clothes and keep the patient warm. Next, try to determine if there is a readily recognizable and potentially reversible cause of the patient’s condition?








