Chapter 61 Severe sepsis
The incidence of sepsis has increased steadily over the last three decades, at least in part because of ageing western populations.1 Nearly 15% of patients in intensive care have severe sepsis and two-thirds of them have septic shock.2,3 Despite our increased understanding, improved support and more powerful antibiotic therapy, severe sepsis is consistently reported as a leading cause of death in non-cardiac intensive care units (ICUs). Mortality remains high3,4 and severe sepsis claims far more lives than the common diseases of western countries such as acute myocardial infarction, stroke and trauma.1,4
DEFINITION
In 1991 the American College of Chest Physicians and Society of Critical Care Medicine convened a consensus conference with the goal of providing a uniform definition for sepsis and its sequelae, using common clinical findings such as alteration in body temperature, tachycardia, tachypnoea and abnormalities in the white blood cell count that were identifiable at the bedside or early in the clinical process.5 They proposed to differentiate systemic inflammation from the inflammatory response itself, and advocated a model in which sepsis is defined as infection in association with a state of systemically activated inflammation (Table 61.1). To differentiate illness severity further, severe sepsis was defined as sepsis with organ dysfunction and septic shock as sepsis with haemodynamic collapse.5
Table 61.1 Sepsis definitions from the 1991 American College of Chest Physician and Society of Critical Care Medicine Consensus Conference5
| Systemic inflammatory response syndrome (SIRS) | At least two among the following: |
| Sepsis | SIRS + confirmed or presumed infection |
| Severe sepsis | Sepsis + organ hypoperfusion or dysfunction |
| Septic shock | Sepsis with refractory hypotension (systolic arterial blood pressure < 90 mmHg, mean arterial pressure < 70 mmHg) or vasopressor dependency after adequate volume resuscitation |
These consensus definitions delineate gradations of mortality risk, but they do not adequately stratify patients into homogeneous groups with respect to the underlying pathophysiology or potential to respond to therapy. They define concepts, but do not identify patients with a single disease.6,7 For these reasons, a new consensus conference held in 2001 presented a new framework of consensus definitions and a staging system. This system, called PIRO (Table 61.2), includes domains for predisposition (premorbid illness), insult/infection (site, bacteriology of infection, severity of other insults such as trauma), response (hypotension, severity of illness measures such as such as Acute Physiology, Age and Chronic Health Evaluation – APACHE), and organ dysfunction (aggregate organ dysfunction scores such as multiple-organ dysfunction syndrome and Sequential Organ Failure Assessment – SOFA).6,7
Table 61.2 The PIRO sepsis staging system6
| Predisposition | Previous illness with reduced probability of short-term survival |
| Age | |
| Genetic polymorphisms in components of inflammatory response | |
| Insult/infection | Culture and sensitivity of infecting pathogens |
| Disease amenable to source control | |
| Gene transcript profiles | |
| Response | Systemic inflammatory response syndrome (SIRS) |
| Sepsis | |
| Severe sepsis | |
| Septic shock | |
| Markers of activated inflammation (C-reactive protein, procalcitonin, interleukin-6) | |
| Markers of impaired host responsiveness (human leukocyte antigen (HLA)-DR) | |
| Detection of therapy target (protein C, tumour necrosis factor, platelet-activating factor) | |
| Organ dysfunction | Number of failing organs |
| Composite scores |
PATHOGENESIS
Severe sepsis and septic shock evolve from a systemic inflammatory and coagulation response to a documented infection. Gram-negative bacilli (mainly Escherichia coli, Klebsiella spp. and Pseudomonas aeruginosa) and Gram-positive cocci (mainly staphylococci and streptococci) are the pathogens most commonly associated with the development of sepsis, although fungi, viruses and parasites cause the syndrome. Fungi, mostly Candida, account for only 5% of all cases of severe sepsis.7,8
The pathophysiology of bacterial sepsis is initiated by the outer-membrane components of both Gram-negative organisms (lipopolysaccharide, lipid A, flagellin and peptidoglycan) and Gram-positive organisms (teichoic acid, lipoteichoic acid, peptidoglycan). These outer-membrane components and other cell wall components are able to bind to the CD14 receptor, a protein anchored in the outer leaflet of the surface of monocytes. Recently, some coreceptors called Toll-like receptors were identified and it was demonstrated that bacterial components also interact with them. Ten members of the Toll-like receptor family have been identified, each of which shows a degree of specificity for pathogenic microorganisms and cellular products (e.g. TLR2 for peptidoglycan, lipoteichoic acid or TLR4 for lipopolysaccharide).7,8
Binding of Toll-like receptors activates intracellular signalling pathways that trigger transcription factors, such as nuclear factor-κB, which in turn control the expression of immune response genes, resulting in the release of cytokines (Figure 61.1). They secrete either cytokines with inflammatory properties (including tumour necrosis factor-α (TNF-α), interleukin-1 (IL-1), IL-2, IL-6) or cytokines with anti-inflammatory properties (IL-4 and IL-10).7,8
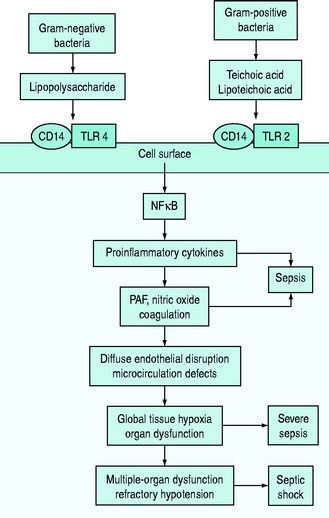
A network of inflammatory mediators activates leukocytes, promotes leukocyte–vascular endothelium adhesion and induces endothelial damage.6 This endothelial damage, in turn, leads to tissue factor expression and activation of the tissue factor-dependent clotting cascade with subsequent formation of thrombin, so that microaggregates of fibrin, platelets, neutrophils and red blood cells impair capillary blood flow, thereby decreasing oxygen and nutrient delivery.6–8
In particular, early inflammatory cytokines increase the expression of the enzyme-inducible nitric oxide synthase (iNOS) in endothelial cells; increased synthesis of the potent vasodilator nitric oxide leads to a decrease in systemic vascular resistance characteristic of shock. Inflammatory cytokines such as TNF also contribute to disruption of the tight junctions between endothelial cells, resulting in increased permeability to plasma proteins and fluid with generalised tissue oedema.7,9 IL-6 alters hepatocyte protein synthesis, inducing the synthesis of acute-phase reactants and promoting anaemia. The acute-phase response also results in downregulation of the production of albumin and anticoagulant proteins such as protein C.7,9 Microcirculatory dysfunction plays a key role in the development of organ dysfunction in septic patients.10 As a result of the vicious cycle of inflammation and coagulation, cardiovascular insufficiency (due to the myocardial-depressant effect of TNF, vasodilation and capillary leak) and multiple-organ failure occur, often leading to death (see Figure 61.1).
DIAGNOSIS
The initial presentation of severe sepsis and septic shock is often non-specific and its severity is cryptic. Diagnosis requires the presence of a presumed or known site of infection, evidence of a systemic inflammatory response syndrome and acute sepsis-associated organ dysfunction (Table 61.3).
Table 61.3 Diagnosis of sepsis-associated organ dysfunctions
| Cardiovascular | Systolic arterial blood pressure < 90 mmHg |
| Decrease in systolic blood pressure > 40 mmHg | |
| Mean arterial blood pressure < 70 mmHg | |
| Decreased capillary refill or mottling | |
| Respiratory | PaO2/Fio2 < 300 |
| Renal | Creatinine increase > 60 μµmol/l from baseline |
| Creatinine increase > 60 μµmol/l within last 24 hours | |
| Urine output < 0.5 ml/kg per hour for 2 hours despite fluid resuscitation | |
| Coagulation | Activated partial thromboplastin time > 60 seconds |
| International normalised ratio > 1.5 | |
| Platelets < 100 000/μµl | |
| Liver | Bilirubin > 70 mmol/l |
| Acid–base | Lactate > 2.1 mmol/l |
Laboratory and invasive haemodynamic measurements include:
CARDIOVASCULAR FAILURE
Cardiovascular failure is caused by an inadequate supply or inappropriate use of metabolic substrate. There is increasing evidence that sepsis is accompanied by a hypermetabolic state, with enhanced glycolysis and hyperlactataemia that do not necessarily indicate tissue hypoxia.11 Hypotension is frequently the result of low systemic vascular resistance. Shock is typically defined as a systolic pressure lower than 90 mmHg that is unresponsive to fluids or that requires vasoactive drugs.
LIVER DYSFUNCTION
The liver is a mechanical and immunologic filter for portal blood and may be a major source of cytokines. An increase in aminotransferase and bilirubin levels is common,12 but hepatic failure is rare.
COAGULATION ABNORMALITIES
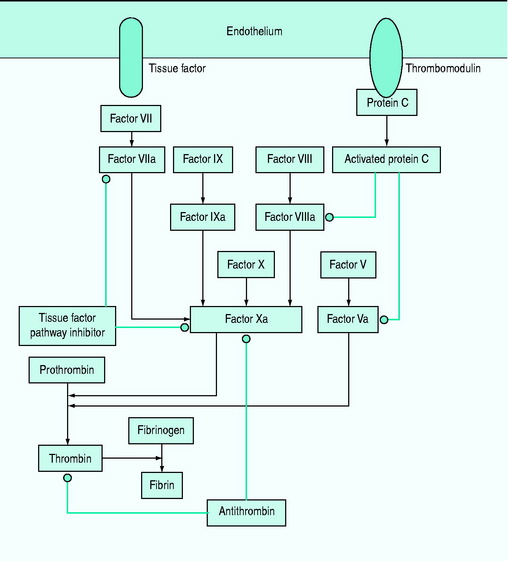
Subclinical coagulopathy, with a mild elevation of the prothrombin or partial thromboplastin time, or a moderate reduction in platelet count or an increase in plasma fibrinogen degradation and D-dimer levels may occur. Coagulopathy is caused by deficiencies of the coagulation system proteins (antithrombin III, protein C, tissue factor pathway inhibitor and the kinin system) (Figure 61.2). Simultaneous activation of coagulation by inflammatory cytokines and reduced production of anticoagulant proteins contribute to disseminated intravascular coagulation.12
SEPSIS-ASSOCIATED ENCEPHALOPATHY
A diffuse cerebral dysfunction is often present in sepsis and may ensue even before signs of other organ failure. It is better defined as ‘sepsis-associated encephalopathy’ (SAE), in order to stress the absence of direct infection of the central nervous system. The main sign of SAE is an altered mental status. Electroencephalography is the most sensitive diagnostic test, and allows the grading of the severity of cerebral dysfunction that is related to outcome. SAE is potentially reversible, but always worsens the prognosis. The pathophysiology of SAE is still not completely understood, and it is probably multifactorial. Indeed, brain dysfunction in sepsis may be related to microorganism toxin activity, to the effects of inflammatory mediators, to metabolic alterations and to abnormalities in cerebral circulation.13,14



Full access? Get Clinical Tree


