▪ TERMINOLOGY
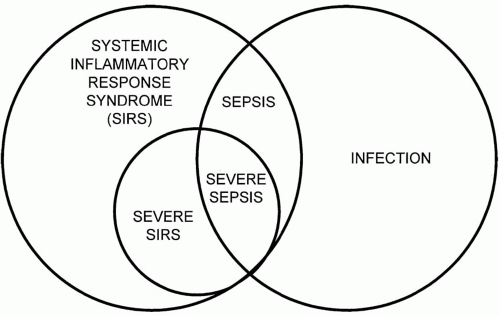
Standardized definitions of three important clinical conditions—systemic inflammatory response syndrome (SIRS), sepsis, and severe sepsis are well accepted. The constellation of fever or hypothermia, tachycardia, tachypnea, and leukocytosis or leukopenia defines SIRS. SIRS represents a body’s response to inflammation that may or may not be due to infection. When a major organ system malfunction results from this process, the syndrome is termed “severe SIRS.” Common parameters for vital sign abnormalities of SIRS are a temperature lower than 96°F (36°C) or higher than 100.4°F (38°C), a pulse greater than 90 to 100 beats/min in the absence of intrinsic heart disease or pharmacotherapy limiting heart rate response, and tachypnea with a respiratory rate higher than 20 breaths/min. In mechanically ventilated patients, a minute volume criterion of 10 L/min is customarily applied. Abnormalities in the circulating leukocyte (WBC) count (>10,000/mm3 or <4,000/mm3) are frequent enough in SIRS to constitute a diagnostic hallmark.
When SIRS is caused by infection, the condition is termed “sepsis.” Correspondingly, when an acute organ system dysfunction occurs in a patient with infection-induced SIRS, the resulting syndrome is called severe sepsis. Hypotensive cardiovascular system failure and those who are normotensive with evidence of systemic hypoperfusion (i.e., lactic acidosis) are said to have “septic shock.” Although strict criteria are lacking, the multiple organ failure syndrome is highly lethal, progressive cumulative failure of organs. The relationship of infection, SIRS, severe SIRS, sepsis, and severe sepsis are illustrated in Figure 27-1. An operational definition of severe sepsis is outlined in
Table 27-1.
▪ EPIDEMIOLOGY
The millions of cases of severe sepsis that occur each year in the world present huge medical, social, and economic problems. Patients average 55 to 60 years of age, and for unclear reasons, there is a male predominance; however, severe sepsis has no age or gender boundaries. With the exception of a spike in frequency in the first year of life, severe
sepsis has a low incidence throughout early adulthood, then an exponentially rising incidence, mortality rate, and cost, after the age of 50. Although sepsis can develop in perfectly healthy persons in the community, most patients have been hospitalized for several days before recognition of the condition. Victims of trauma, immunosuppressed patients, and patients with chronic debilitating medical conditions (e.g., diabetes, chronic obstructive lung disease) or undergoing complicated surgical procedures are most at risk.
Overall, 30% to 40% of patients with severe sepsis die despite receiving “standard therapy” consisting of antimicrobial therapy and organ system support with fluids, vasoactive drugs, mechanical ventilation, dialysis, and nutrition therapy. Elderly and hypothermic patients have a substantially worse prognosis than those without these factors; however, the best practical clinical predictor of outcome is the number of dysfunctional organ systems. Among the possible organ failures, circulatory failure (shock) has a disproportionate negative prognostic value. The morbidity and mortality of severe sepsis remain unacceptably high, and billions of dollars are spent caring for this desperately ill group of patients. Fortunately, survivors usually eventually regain premorbid levels of function in most organs; however, there is a growing awareness that a significant proportion of patients are left with long-lasting cognitive and neuromuscular impairment.
The average survivor requires 7 to 14 days of intensive care support, with much of this time spent on a ventilator. After intensive care unit (ICU) discharge, an additional 10- to 14-day hospital stay is typical. Thus, the hospital length of stay for survivors averages 3 to 5 weeks. After hospital discharge, long-term skilled inpatient care or challenging home care is often required. Most survivors are discharged on numerous medications; see physicians frequently during the year after discharge and are readmitted one or more times for treatment of complications. Many are shocked to learn that severe sepsis victims typically generate a hospital bill of $150,000 to $200,000 and million dollar bills are not unheard of.
▪ RELATIONSHIP OF INFECTION TO SEPSIS
Recovery of a pure growth of a pathogen from a normally sterile site (e.g., blood or joint or cerebrospinal fluid [CSF]) diagnoses
infection; however, most infected patients do not develop severe sepsis.
This fact suggests that it is not infection per se that is etiologic but rather the combination of infection and host response that determines if an individual will develop severe sepsis. Interestingly, a clear microbiologic explanation is absent in many patients even though cultures grow some organism 60% to 80% of the time. Many of these “positive” cultures are obtained long after severe sepsis is established and represent insignificant colonization, contamination, or superinfection. Common examples include growth of skin flora in one of several blood culture bottles, the recovery of a light growth of
Staphylococcus aureus from sputum of a ventilated patient, or demonstration of a few colonies of
Candida albicans in the urine of a patient with an indwelling urinary catheter. Perhaps the most convincing evidence of infection comes when several blood cultures obtained at the onset of the episode grow an identical pathogen consistent with the patient’s clinical situation, for example, recovery of
Escherichia coli in multiple blood cultures from an elderly man with bladder outlet obstruction and pyuria. Unfortunately, positive blood cultures are seen in no more than 20% to 30% of severe sepsis patients, and blood cultures are very rarely (perhaps as low as 1%) positive if obtained after antimicrobial therapy is started. Despite historical teaching, there is little prognostic import of having positive blood cultures, unless bacteremia cannot be eradicated. Inability to clear the circulation of organisms is often associated with an unresolved focus of infection (e.g., endocarditis or an infected foreign object) and portends a worse prognosis.
Remarkably, the severity of the host response depends little on the presence of infection. In fact, noninfected patients with severe SIRS caused by pancreatitis, trauma, or burns have similar biochemical changes and identical physiology, clinical presentation, and outcome as infected patients with severe sepsis. Again, this observation suggests that infection is not essential but rather that microbiologic stimulation acts merely as one disease trigger.
Consistently, the lung is the most common site of infection leading to severe sepsis accounting for roughly half of all the cases. Intra-abdominal infections (20% to 25%) and urinary tract infections (approx. 10%) are the next most common, with all other sites comprising the remaining 15% of infections. Despite these statistics, in an individual, any site of infection may occur, thus a thoughtful assessment must be conducted every time severe sepsis develops.
▪ MICROBIOLOGY
Bacteria, fungi, parasites, and viruses all can incite severe sepsis. Probably, because of the relatively high incidence of bacterial infections and relative ease in recovery of the organisms, bacteria are most commonly implicated. Limitations of diagnostic techniques make viruses least commonly identified. Historically, fungal infections were rarely etiologic in immunocompetent hosts but with improved antibacterial agents and support techniques, more and more immunocompetent patients are surviving long enough to acquire a fungal infection. Distressingly, the frequency of fungal, particularly Candidal, infection is rising dramatically and now accounts for almost 10% of all episodes of severe sepsis.
When a bacterial pathogen is identified, the frequency of Gram-positive versus Gram-negative bacteria is roughly 50:50. Discussions of the likelihood of Gram-positive versus Gram-negative infection are of limited value; the prevalence of organisms varies by location and over time, “cycling” under antibiotic pressure. Furthermore, knowing the frequency of each type of bacteria in a population is not particularly helpful in designing therapy for one patient except that it can highlight unusual local resistance patterns. For example, in some parts of the south-eastern United States half of all Pneumococcus isolates are at least of intermediate resistance to penicillin. In addition, now in some ICUs the single most common organism causing severe sepsis is a highly resistant nosocomial pathogen, typically methicillin-resistant S. aureus (MRSA) or vancomycin-resistant Enterococcus. Regardless, in most circumstances, critically ill patients require prompt empiric therapy for all reasonably likely organisms until culture data are available.
▪ PATHOPHYSIOLOGY
The severity of severe sepsis is determined more by the specificity and ferocity of the host response than by the inciting organism. Ironically, the same inflammatory and coagulopathic mechanisms that are detrimental when undirected in the septic patient are probably beneficial on the average day. Certainly, both confined inflammation and accelerated coagulation are beneficial when they limit spread of local infection or injury. It is only when rogue, diffuse, unbridled inflammation or coagulation occurs that they are detrimental.
Historically, excessive inflammation was considered the major, if not sole, pathogenetic factor in
severe sepsis. This paradigm envisioned a multistage inflammatory “cascade” in which an initial trigger caused production of a few “early” mediators, followed over hours by a larger number of secondary mediators (
Table 27-2). It is now clear that inflammation is but one of at least three important pathophysiologic pathways that also includes enhanced coagulation and impaired thrombolysis.
The trigger for severe sepsis is often a protein, lipid, or carbohydrate toxin shed from a microbe but may be activated complement, a clotting cascade component, or dead host tissue. The most notorious inciting toxin is endotoxin, the integral cell wall lipopolysaccharide component of Gramnegative bacteria. However, it is far from being the only important toxin; staphylococcal toxic shock syndrome toxin (TSST-1) and group B streptococcal (GBS) toxin are other well-recognized triggers. The triggering compound usually is only present transiently in the circulation and commonly escapes detection, even when sophisticated monitoring is performed. For example, less than one half of patients exhibiting septic shock ever have detectable endotoxin in plasma. This fact may help to explain the failure of antidotes developed to bind and neutralize circulating toxins. Development of sepsis does not require bacteremia or endovascular infection; toxic products may be released into the bloodstream from localized sites (e.g., abscesses) or directly from the colon (gut translocation), even when viable organisms do not circulate.
Tumor necrosis factor (TNF) and interleukin 1 (IL-1) have received the most attention as targets for modifying the septic response because they are potent, rapidly produced inflammatory compounds found in the tissues and circulation of many septic patients (
Table 27-3). However, controversy exists regarding the significance of circulating cytokines, and clinical trials designed to lessen levels of these compounds have not reduced mortality. That controversy not withstanding, these cytokines are major stimulants of other mediators, including IL-6, IL-8, enzymes, prostaglandins, leukotrienes, oxidant radicals, platelet-activating factor, and nitric oxide and activate coagulation.
There is growing appreciation that abnormal coagulation is nearly universal in severe sepsis and that a complex interplay exists between clotting and inflammation. At the outset of the syndrome, tissue factor expressed by leukocytes and endothelium and cytokines lead to the production of thrombin by stimulating clotting factors V and VIII. Initially, the natural anticlotting systems (e.g., protein C, protein S, antithrombin) counteract the accelerated clotting. In this process, clotting proteins are consumed forming thrombi, and anticlotting proteins are depleted trying to inhibit clot formation. Because sepsis also impairs the host’s
ability to convert inactive anticlotting precursors to functioning proteins, clotting proceeds unopposed. As a second line of defense, endogenous fibrinolytic systems (e.g., plasminogen) are activated to dissolve the microvessel clogging thrombi, increasing plasma levels of clot degradation products. Although routine clotting assays (e.g., prothrombin and activated partial thromboplastin times) may be near normal, abnormalities of clotting and anticlotting systems can be detected using more sensitive laboratory tests. For example, essentially all patients have elevated fibrin degradation products (e.g., d-dimers) and the vast majority have depleted levels of specific clotting and anticlotting proteins. Unfortunately, thrombin also augments levels of plasminogen activator inhibitor 1 (PAI-1) and thrombin-activatable fibrinolysis inhibitor (TAFI), both antagonists of endogenous thrombolysis. Together, the net effect of PAI-1 and TAfiis to stabilize whatever thrombi form impairing tissue perfusion. Finally, thrombin independently promotes adherence of leukocytes to endothelial cells leading to vessel wall damage. The complex interrelationship of the three major pathways (inflammation, coagulation, and fibrinolysis) is illustrated in Figure 27-2.
Only gold members can continue reading.
Log In or
Register to continue
Related

Full access? Get Clinical Tree


 , O
, O






