(1)
Division of Pulmonary and Critical Care Medicine, Eastern Virginia Medical School, Norfolk, VA, USA
Keywords
SepsisSeptic shockHemodynamicsFluid responsivenessAntibioticsPathophysiologySystemic inflammatory response syndrome (SIRS)Organ dysfunctionManagementLactateEarly goal directed therapy (EGDT)Surviving Sepsis Campaign (SSC)Except for a few occasions patients’ appear to die from the body’s response to infection rather than from it
William Osler, Physician (1849–1919)
The word “sepsis” is derived from the ancient Greek word for rotten flesh and putrefaction. Since then, a wide variety of definitions have been applied to sepsis, including sepsis syndrome, severe sepsis, bacteremia, septicemia and septic shock In 1991, the American College of Chest Physicians (ACCP) and the Society of Critical Care Medicine (SCCM) developed a new set of terms and definitions to define sepsis in a more “precise manner” [1, 2]. These definitions take into account the findings that sepsis may result from a multitude of infectious agents and microbial mediators and may not be associated with detectable bloodstream infection. The term “systemic inflammatory response syndrome” (SIRS) was coined to describe the common systemic response to a wide variety of insults. When SIRS was the result of a suspected or confirmed infectious process, it is termed “sepsis”. Severe sepsis was defined as sepsis plus organ dysfunction. Septic shock is a subset of severe sepsis and was defined as “sepsis-induced hypotension persisting despite adequate fluid resuscitation” (see Fig. 12.1). While the quantity of fluid that qualifies as “adequate fluid resuscitation” is controversial, we believe septic shock is best defined as a “mean arterial pressure (MAP) less than 65 mmHg after a fluid challenge of 20 mL/Kg body weight (given 30–60 minutes) in patients with sepsis and in the absence of other causes for hypotension” (also see Chap. 14). According to the ACCP/SCCM definitions, three stages in the hierarchy of the host’s response to infection were recognized, namely, sepsis, severe sepsis and septic shock, with sepsis having the best prognosis and septic shock the worst. While the use of the SIRS criteria to define sepsis is somewhat controversial [3–6], many consider sepsis to be best defined as the “systemic response to infection with the presence of some degree of organ dysfunction” [5].
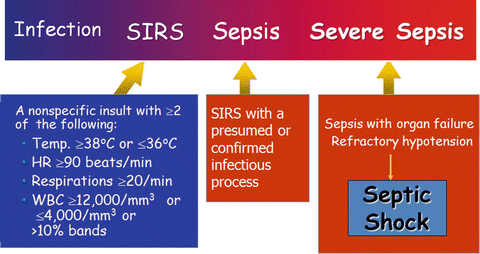
Fig. 12.1
Sepsis: defining a disease continuum
Sepsis is amongst the most common reason for admission to ICU’s throughout the world. An epidemiologic study in European ICU’s demonstrated an incidence of 37 % for sepsis and 30 % for severe sepsis [7]. While the exact incidence of sepsis in the US is unclear (300–1,000 cases/100,000 population per year) the annualized incidence has been reported to have increased by between 8.7 to 13 % over the last 30 years [8–11]. The aging of the population is believed to be largely responsible for the increasing incidence of sepsis in developed counties [12]. Epidemiological data from 2004 to 2009 demonstrated a decrease in in-hospital mortality from 35 to 26 % [8]. This study estimated that there were 229,044 deaths from severe sepsis in 2009, which would place severe sepsis as the third most common cause of death in the US, after heart disease and malignant neoplasms [8]. The 1-year all-cause mortality of patients treated for severe sepsis and septic shock maybe as high as 44 % [13]. In addition, recent data suggests that patients who have had a septic episode are at an increased risk of death for up to 5 years following the acute event [14, 15]. Sepsis is reported to be more common in men and among nonwhite persons [10]. Surgical patients account for nearly one third of sepsis cases in the US; this is important as the management of “surgical” and “medical sepsis” differs somewhat [10].
Bacteriology and Sites of Infection
Bacteriologic data from sepsis trials published during the last two decades indicate the following pattern of culture results:
Gram-positives 25 %
Gram-negatives 25 %
Gram-positive + Gram-negative organisms 15 %
Fungal pathogens 3–5 %
Anaerobes 2 %
Culture −ve 25 %
The most common sites of infection are:
Lung 50 %
Abdomen/pelvis 25 %
Primary bacteremia 15 %
Urosepsis 10 %
Vasculare access 5 %
Pathophysiology of Sepsis
Sepsis is an exceedingly complex condition. Exposure of human macrophages to bacterial antigens has been demonstrated to result in a significant change in the expression over 950 genes [16]. These include genes for pro and anti-inflammatory cytokines, chemokines, adhesion molecules, transcription factors, enzymes, clotting factors, stress proteins and anti-apoptotic molecules. These gene products alter the function of every cell and tissue in the body. Furthermore these mediators interact in complex positive and negative feedback loops and result in epigenetic modifications which further alter the expression of this network of mediators [17]. The early phase of sepsis is generally believed to result from the uncontrolled production of pro-inflammatory mediators the so-called “cytokine storm” [18]. However, recent data suggests that both a pro-inflammatory and an opposing anti-inflammatory response occur concurrently in patients with sepsis [19, 20]. In general, following a variable time course, patients transition from a predominantly pro-inflammatory to an anti-inflammatory immunosuppressive state [17–21].
The pathogenetic mechanism and physiological changes associated with sepsis are exceedingly complex with our understanding of this topic rapidly evolving; the reader is referred to excellent reviews on this topic [17–21]. The major pathophysiologic changes in patients with severe sepsis and septic shock include vasoplegic shock (distributive shock), myocardial dysfunction, altered microvascular flow, activation of coagulation and a diffuse endothelial injury [22, 23]. These pathophysiologic changes play a central role in the early management of patients with sepsis. The widespread endothelial injury results in a microvascular leak with tissue and organ edema, hypotension and shock. Increased endothelial permeability is caused by shedding of the endothelial glycocalyx and the development of gaps between endothelial cells (paracellular leak) [24, 25]. Vasoplegic shock, due to failure of the vascular smooth muscle to constrict, results in arterial and veno-dilatation [22]. Vasoplegic shock is believed to be due to increased expression of inducible nitric oxide synthetase with increased production of nitric oxide (NO), activation of K ATP channels resulting in hyperpolarization of the muscle cell membrane, increased production of natriuretic peptides (which act synergistically with NO) and a relative vasopressin deficiency [22]. Veno-dilatation increases the size of the non-stressed blood volume decreasing venous return which compounds the intra-vascular volume deficit caused by the vascular leak.
Septic “Cardiomyopathy”
Reversible myocardial depression in patients with septic shock was first described in 1984 by Parker et al. using radionuclide cineangiography [26]. In a series of 20 patients, they reported a 50 % incidence of left ventricular (LV) systolic dysfunction, defined by an ejection fraction <40 %. Paradoxically, in this study the initial ejection fraction and ventricular volumes were normal in non-survivors and these indices did not change during serial studies. The authors suggested that survival depends on the ability of the left ventricle to dilate and increase stroke volume. It is noteworthy that the “septic cardiomyopathy” is acute and reversible. In a series of 90 patients with severe sepsis, Jardin et al. reported that the left ventricular ejection fraction (LVEF) was depressed in all patients and normalized over a few days in the survivors [27]. In this study, LVEF was significantly lower in survivors than non-survivors, while the LVEDV was significantly smaller in non-survivors than survivors.
The initial studies evaluating cardiac function in sepsis focused on LV systolic function. However, left ventricular diastolic dysfunction has emerged as common finding in patients with severe sepsis and septic shock. Adequate filling during diastole is a crucial component of effective ventricular pump function. Diastolic dysfunction refers to the presence of an abnormal left-ventricular diastolic distensibility, filling, or relaxation, regardless of LVEF. Unlike systolic LV dysfunction, diastolic dysfunction appears to be an important prognostic marker in patients with sepsis. Impaired diastolic distensibility probably accounts for the reduced LVEDV in the studies by Parker and Jardin (referenced above). The reported incidence of systolic and diastolic function in patients with sepsis varies widely; this may be related to the population studied and the diagnostic strategies used. This data is summarized in Table 12.1. It is noteworthy that diastolic dysfunction appears to be more common than systolic dysfunction and that isolated diastolic dysfunction was present in 21 % of patients. Combined systolic and diastolic dysfunction was reported in 16 % of patients. The optimal approach to the management of septic patients with diastolic dysfunction likely differs from that of patients with systolic (or combined) left ventricular dysfunction. As diastolic dysfunction is best diagnosed by Tissue Doppler Imaging (TDI), patients with sepsis who are admitted to the ICU should undergo both bed-side ECHO as well as a formal echocardiographic study. Patients who have a hyperdynamic circulation on bedside ECHO, with a low stroke volume (as determined by non-minimally invasive cardiac output monitoring) and who are volume non-responders should be suspected of having predominant diastolic LV dysfunction (see case example at end of this Chapter).
Table 12.1
Incidence of systolic and diastolic dysfunction in patients with sepsis
Author | Year | n | Systolic dysfunction (%) | Diastolic dysfunction (%) | Isolated diastolic dysfunction% |
|---|---|---|---|---|---|
Parker [26] | 1984 | 20 | 50 | – | – |
Jardin [28] | 1990 | 21 | 29 | – | – |
Etchecopar-Chevreuil [29] | 2008 | 35 | 46 | 20 | 9 |
Veillard-Baron [30] | 2008 | 67 | 60 | – | – |
Bouhemad [31] | 2008 | 54 | 20 | 40 | 20 |
Bouhemad [32] | 2009 | 45 | 18 | 18 | – |
Sturgess [33] | 2010 | 21 | 67 | 57 | – |
Turner [34] | 2011 | 153 | 32 | – | – |
Landesberg [35] | 2012 | 262 | 23 | 54 | 40 |
Pulido [36] | 2012 | 106 | 27 | 37 | 27 |
Mahjoub [37] | 2012 | 83 | – | 57 | – |
Brown [38] | 2012 | 78 | 26 | 61 | 23 |
Mourad [39] | 2014 | 72 | 54 | 46 | 16 |
Summary | 1,038 | 33 | 47 | 23 |
Many factors may contribute to cardiac dysfunction during sepsis. Studies performed in humans have ruled out coronary hypoperfusion as a cause of LV dysfunction in sepsis. Cytokines, particularly TNF-α have been postulated to play a major role in the genesis of septic cardiomyopathy. It has been suggested that the effect of cytokines on cardiac myocytes results from an increase in intracellular nitric oxide and cGMP [40]. Tavernier et al. suggested that increased phosphorylation of troponin I was involved in the septic cardiomyopathy by reducing myofilament response to Ca2+ [41]. Many other additional mechanisms have been suggested including myocardial apoptosis, altered myocardial microcirculation, autonomic neuropathy, mitochondrial dysfunction, abnormal energy metabolism, abnormal adrenergic signalling, and myocardial edema, to name but a few [42–45]. It is unclear why some patients develop systolic LV dysfunction while others develop diastolic dysfunction. However, older age, diabetes, hypertension and obesity may increase the likelihood of developing diastolic LV dysfunction [35, 36]. Furthermore, aggressive fluid resuscitation may increase myocardial edema increasing the risk of diastolic dysfunction [33].
Complications Associated with Sepsis
Complications associated with severe sepsis and septic shock includes acute respiratory distress syndrome (ARDS), acute kidney injury (AKI), disseminated intravascular coagulation (DIC), critical illness polyneuropathy and “septic encephalopathy”. The risk of ARDS (and outcome) may be critically dependent on the fluid resuscitation strategy (see below). Sepsis is the leading cause of AKI in critically ill patients and is associated with a hospital mortality exceeding 50 % [46, 47]. Contrary to “classic teaching,” animal models and human studies have shown that the sepsis-induced AKI occurs in the setting of preserved or increased renal blood flow [48]. A “unifying theory” has been proposed to explain the development of sepsis-induced AKI which includes the interplay between inflammation and oxidative stress, microvascular dysfunction, and the adaptive response of the tubular epithelial cell to the septic insult [48]. Activation of the coagulation cascade with the generation of fibrin is a pathologic hallmark of sepsis. Activation of coagulation during sepsis is primarily driven by the tissue factor (TF) pathway. Due to activation of the coagulation cascade almost all septic patients are thrombocytopenic (or have a falling platelet count), and indeed a normal platelet count makes the diagnosis of sepsis unlikely. An elevated d-dimer, thrombin-antithrombin complexes and a prolonged prothrombin time are found in the majority of patients with severe sepsis while anti-thrombin, protein C and protein S levels are significantly decreased.
Clinical Features and Diagnosis of Sepsis
Sepsis is a systemic disease with a variety of clinical manifestations. The initial symptoms of sepsis are non-specific and include malaise, tachycardia, tachypnea, fever and sometimes hypothermia. Although most patients with sepsis have an elevated white cell count, some patients present with a low white cell count, which in general, is a poor prognostic sign. A band count in excess of 10 % has been reported to have a high specificity (92 %) but low sensitivity for the diagnosis of sepsis (43 %) [49] Other clinical manifestations include altered mental status, hypotension, respiratory alkalosis, metabolic acidosis, hypoxemia with acute lung injury, thrombocytopenia, consumptive coagulopathy, proteinuria, acute tubular necrosis, intra-hepatic cholestasis, elevated transaminases, hyperglycemia and hypoglycemia. Patients may present with clinical features of a localized site of infection, such as cough, tachypnea and sputum production due to pneumonia; flank pain and dysuria with urinary tract infection and abdominal pain with intra-abdominal infection.
Organ Dysfunction in Severe Sepsis/Septic Shock
Cardiovascular
Tachycardia
Hypotension
Decreased contractility
Vasodilatation
Respiratory
Tachypnea
Decreased PaO2/FiO2
ALI/ARDS
Hematological
Thrombocytopenia
Increased PTT/PT
Decreased protein C
Increased d-dimer
Neurological
Confusion
Agitation
Altered consciousness
Neuropathy
Myopathy
Cerebral edema
Renal
Oliguria
Increased s-creatinine
Hepatic
Increase in transaminases
Decreased albumin
Metabolic/endocrine
AG acidosis
Increased lactate
CIRCI (adrenal insufficiency)
Hyperglycemia/hypoglycemia
Hypophosphatemia
The manifestations of sepsis can sometimes be quite subtle, particularly in the very young, the elderly and those patients with chronic debilitating or immunosuppressing conditions. These patients may present with normothermia or hypothermia. The failure to generate a temperature greater than 99.6 °F (37.5 °C) in the first 24 h of clinical illness, has been associated with an increased mortality rate. An altered mental state or an otherwise unexplained respiratory alkalosis may be the presenting feature of sepsis.
The signs and symptoms of systemic inflammation are not useful in distinguishing infectious from non-infectious causes of SIRS. Despite exhaustive microbiological tests a pathogen is not isolated in about 25 % of patients [50]. Blood cultures are considered to provide the clinical gold standard for the diagnosis of bacterial infections. However, blood cultures are only positive in between 20 and 30 % of patients with sepsis; moreover, it takes 2–3 days before the results become available. Molecular methods based on polymerase chain reaction (PCR) technology hold promise for the early diagnosis of bacterial infection and for pathogen identification. A number of bio-markers have been evaluated as more specific indicators of infection.
Procalcitonin (PCT) has to date been the most useful bio-marker to aid in the diagnosis of sepsis. PCT, a propeptide of calcitonin, is normally produced in the C-cells of the thyroid. In healthy individuals, PCT levels are very low (<0.01 ng/mL). In patients with sepsis, however, PCT levels increase dramatically, sometimes to more than several hundred nanograms per milliliter. The use of PCT for the diagnosis of sepsis and in determining the duration of antibiotics is controversial. The test is not perfect and should always be interpreted in the clinical context together with other diagnostic tests. Su et al. evaluated the diagnostic accuracy of 32 clinical signs, symptoms and laboratory tests available during a patients stay in the Emergency Department [51]. In this study, PCT was that variable that had the best diagnostic accuracy. Similarly, Tromp et al. demonstrated that PCT had the best predictive value for bacteremia with an area under the receiver operator curve (ROC) curve of 0.80, with a sensitivity of 89 % and a specificity of 58 % [52]. In this study, the predictive value of a combination of PCT plus a panel of other biomarkers, clinical signs, or analysis of serial PCT levels did not lead to a significant improvement of the predictive value of PCT alone. Wacker performed a meta-analysis to evaluate the diagnostic accuracy of PCT [53]. In this meta-analysis the sensitivity was 0.77 (95 % CI 0.72–0.81), the specificity was 0.79 (95 % CI 0.74–0.84) and the area under the ROC curve was 0.85 (95 % CI 0.81–0.88). This diagnostic accuracy is better than any other single test to diagnose sepsis. A PCT >0.5 ng/mL is highly suggestive of a bacterial infection while a level <0.1 ng/mL makes this diagnosis less likely [54]. However, the optimal diagnostic threshold is unclear and has been reported to vary from 0.25 to 1.4 ng/mL [52, 54]. This variation in diagnostic threshold may partly be explained by the case-mix of each study and the fact that patients’ with gram-negative infection have significantly higher PCT levels than those with gram-positive infections [55–57]. Infection with a gram negative pathogen is highly likely in a patient with a PCT level >5 ng/mL. It should be noted that patients with fungal infections usually have much lower or “normal” PCT level [55]. In hematologic patients an elevated PCT level within 24 h after the onset of neutropenic fever is highly predictive of Gram-negative bacteremia [58]. In addition to being a very useful test to diagnose bacterial sepsis, the trend in the PCT level is useful for deciding when to discontinue antibiotics [59, 60]. Furthermore, the trend in the PCT is strongly predictive of outcome, with a persistently high level being associated with a poor outcome [61].

Don’t Miss the Diagnosis of Sepsis
In many patients who present to the ED the diagnosis of sepsis is obvious, i.e. high fever, high WBC and an obvious source of infection. However, in many instances the patient may present with vague constitutional symptoms, mild hypotension and tachycardia or with a fever and myalgia that are attributed to “a viral syndrome”. DO NOT send these patients home without further workup (unless they obviously have a viral syndrome); YOU DO NOT want to send the patient home, who then returns some time later in septic shock, and subsequently loses an arm or leg (or both) or dies. When the diagnosis is not clear you must do a WBC with differential and band count, blood lactate level and PCT.
Any one of the following features alone or in combination are suggestive of bacterial sepsis:
Fever >38.3 °C
Heart rate >120/min
Systolic BP <90 mmHg
PCT >0.5 ng/mL
Bandemia >5 %
Lymphocytopenia <0.5 × 103ul
Thrombocytopenia <150 × 103uL
Lactate >2.0 meq/L
Increased neutrophil/lymphocyte ratio
Management of Sepsis
On the 8th November 2001 Emanuel Rivers and collaborators published a study entitled “Early Goal Directed Therapy in the treatment of severe sepsis and septic shock” in which they compared two protocols for the early resuscitation of patients with severe sepsis and septic shock (for 6 h in the Emergency Department) [62]. Both protocols used the CVP to guide fluid therapy. The “treatment arm” (EGDT) required placement of an oximetric central venous catheter with protocolized interventions to maintain the saturation of the central venous oxygen saturation (ScvO2) >70 % (see Fig. 12.2). The study which enrolled 288 patients (25 were excluded after the fact) and reported a 28 day mortality of 49.2 % in the control group and 33.3 % in the EGDT group (p = 0.01, with an absolute reduction in the risk of death of 16 %). A number of serious concerns have been raised with regards to this study including the validity of the protocol itself, the conduct of the study and the reporting of the study [63–68]. These include:
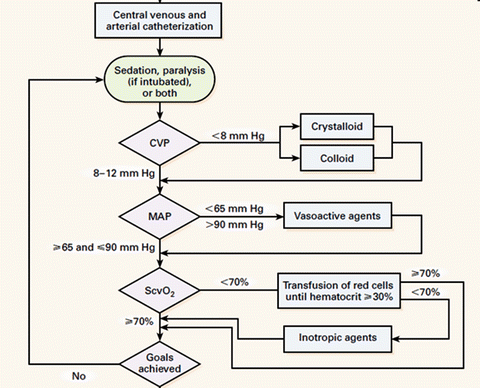
Fig. 12.2
The “infamous” River’s Early Goal Directed Therapy (EGDT) protocol
i.
The results of the study appear to be biologically implausible
ii.
The study was not blinded, with concerns regarding interference with the conduct of the study
iii.
None of the elements of the protocol were (are) supported by any level of credible scientific evidence
iv.
The mortality in the control group was significantly higher than that of any study reported at that time [69], raising the question of sub-standard care in the control arm of the study
v.
The hemodynamic profile of the patients, particularly the low ScvO2 (49 ± 13 %) and the high MAP (76 ± 24 mmHg), is very atypical for patients with sepsis
vi.
The senior author had serious undisclosed conflicts of interest.
vii.
Twenty five patients were excluded from the final analysis (for unclear reasons). When these patients are included in an intention-to-treat analysis it has been reported that “the death gap was no longer statistically significant.” [63]
Despite the limitations of this single center study, EGDT soon became regarded as the “standard of care” around the world, it was endorsed by major organizations in the US (including the Institute for Health Care Improvement (IHI), Centers for Medicare and Medicaid Services (CMS), Agency for Health Care Quality and Improvement (AHQI), Society of Critical Care Medicine) and abroad and formed the basis of the 6-h resuscitation bundle of the 2004, 2008 and 2012 recommendations of the Surviving Sepsis Campaign (SSC) [70–72]. A large number of before-and-after studies (probably >30 studies) have been published claiming that the use of the Surviving Sepsis Campaign 6-h resuscitation bundle reduced mortality (see Chap. 1 on the limitations of before-after studies) [73, 74]. However, it is noteworthy that the reported mortality has fallen when compliance with “the bundle” is less than 10–15 %, but has increased when compliance with the bundle is high [75–77]. Shiramizo et al. noted a fall in mortality in their patients with severe sepsis/septic shock from 41.4 to 16.2 % between 2008 and 2009 despite a decline in compliance with the 6-h resuscitation bundle from 21.1 to 13.7 % [78]. We postulate that the improved outcome with low “bundle” compliance is related to improved screening for sepsis and earlier administration of antibiotics with concomitant poor achievement of the CVP targets, while the increased mortality with high compliance of the “bundles” is related to the harm of achieving the CVP targets i.e. the benefit of early antibiotics are outweighed by the harm of achieving the CVP targets (see harms of excess fluid and high CVP) [73, 74]. This postulate is supported by the analysis of Barochia et al. who concluded that “only antibiotics meet the stated criteria of proof for bundle inclusion” [79].
In 2014, 13 years after the publication of the EGDT study the ProCESS study was published [80]. Process enrolled 1,341 patients, of whom 439 were randomly assigned to protocol-based EGDT (Rivers EGDT), 446 to protocol-based standard therapy, and 456 to usual care. There was no significant difference in 90-day and 1 year mortality between groups. However, in the sickest sub-group of patients (those with a baseline lactate >5.3 mmol/L) the mortality was significantly higher in the EGDT group as compared to usual care (38.2 vs. 26.4; p = 0.05). ProCESS has now clearly established that EGDT should be abandoned. This does not mean that patients with sepsis should not be managed by evidence based guidelines or recommendations. Unfortunately, a rational approach to the management of sepsis has been contaminated by the Rivers EGDT protocol and the Surviving Sepsis Campaign.
This section will focus on those interventions which are currently believed to improve the outcome of patients with severe sepsis and septic shock. Experimental immunomodulating interventions including the use of novel molecules and antibodies [17], extracorporeal blood purification [81] or approaches to “seal” the leaky endothelium [25] will not be discussed. The early management of patients with sepsis centers on the administration of antibiotics, limited amounts of intravenous fluids and vaso-active agents, followed by source control. Unfortunately, there is no high quality evidence (from one or more randomized controlled trials) demonstrating that any of these interventions alter outcome. It is however likely that the early detection of sepsis with the timely administration of appropriate antibiotics is the single most important factor in reducing the morbidity and mortality from sepsis [79]. It has become increasingly apparent that in many patients there is a long delay in both the recognition of sepsis and the initiation of appropriate therapy. This has been demonstrated to translate into an increased incidence of progressive organ failure and a higher mortality [75, 82]. Clinicians therefore need to have a high index of suspicion for the presence of sepsis. The clinical features which should heighten the index of suspicion for the diagnosis of sepsis are listed above.
Antibiotic Therapy
Empiric intravenous antibiotic therapy should be started as soon as possible and within the first hour of recognition of severe sepsis, after appropriate cultures have been obtained. In a retrospective analysis of 2,600 patients, Kumar and colleagues demonstrated that the risk of dying increased progressively with an increase in the time to receipt of the first dose of antibiotic from the onset of sepsis induced hypotension [83]. Furthermore, there was a 5–15 % decrease in survival with every hour delay over the first 6 h. The choice of antibiotics is largely determined by the source or focus of infection, the patient’s immunologic status and severity of infection, the risk of infection with a multi-drug resistant pathogen (see Chap. 17: The Pneumonias) as well as knowledge of the local microbiology and sensitivity patterns. Initial empirical anti-infective therapy should include one or more drugs that have activity against the likely pathogens and that penetrate into the presumed source of sepsis. Because the identity of the infecting pathogen(s) and its sensitivity pattern(s) are unknown at the time of initiation of antibiotics, in patients with severe sepsis and septic shock the initial regimen should include two or more antibiotics or an extended spectrum β-lactam antibiotic with the aim of treating all realistically possible microbial causes (see also Chap. 17). A number of studies have demonstrated that appropriate initial antimicrobial therapy, defined as the use of at least one antibiotic active in vitro against the causative bacteria reduced mortality when compared with patients receiving inappropriate therapy [84–86]. Once a pathogen is isolated, monotherapy is adequate for most infections; this strategy of initiating broad spectrum cover with two or more antibiotics and then narrowing the spectrum to a single agent when a pathogen is identified is known as “antimicrobial de-escalation.”[87] Antimicrobial de-escalation has been demonstrated to be associated with a reduction in hospital mortality [88]. The indications for continuation of double-antimicrobial therapy include enterococcal infections and severe intra-abdominal infections. In addition, double antimicrobial therapy (3rd generation cephalosporin and macrolide) is recommended for patients with severe community acquired pneumonia and those with pneumococcal bacteremia (see Chap. 17) [89–91]. In order to rapidly achieve adequate blood and tissue concentrations, antibiotics should be given intravenously, at least initially. Dosing regimens should take into account whether the antibiotic “kills” by time-dependent kinetics (e.g. B-lactam antibiotics, vancomycin) or concentration-dependent kinetics (e.g. aminoglycoside) [85, 86, 92, 93]. The clinical effectiveness of β-lactam antibiotics and vancomycin is optimal when the concentration of the antimicrobial agent in the serum exceeds the minimum inhibitory concentration (MIC) of the infecting organism for at least 40 % of the dosing interval. In addition, antibiotic dosing should also take into account the patient’s hepatic and renal function.
Fluid Therapy (See also Chap. 9)
Beyond the early administration of antibiotics, aggressive “supportive measures” may be harmful and the “less is more” paradigm appears applicable for the management of patients with severe sepsis. In these highly vulnerable patients, more intensive treatment may promote the chances of unwanted adverse effects and hence, iatrogenic injury [94]. Current teaching suggests that aggressive fluid resuscitation is the best initial approach for the cardiovascular instability of sepsis. Consequently large volumes of fluid (5–10 L) are often infused in the early stages of sepsis. There is however no human data that substantial (>30 mL/kg) fluid resuscitation reliably improves blood pressure or end-organ perfusion [95, 96]. This approach is likely to lead to “iatrogenic salt water drowning” with severe ARDS, AKI and death.
From a pathophysiological point of view, large volume fluid resuscitation in patients with sepsis is illogical and may worsen the hemodynamic derangements of sepsis. In patients with septic shock who are fluid responders (an increase in cardiac output with fluid boluses) vasodilatation with a fall in systematic vascular resistance has been observed following fluid resuscitation [97, 98]. A similar finding has been noted in an experimental sepsis model [99]. Hence, although the cardiac output increases, vasodilatation occurs and the blood pressure may remain unchanged [97]. Increased vascular shear stress consequent to the increase in cardiac output increases the expression of nitric oxide synthetase with increased release of nitric oxide [22]. In addition, increased cardiac filling pressures increase the release of natriuretic peptides which act synergistically with nitric oxide causing cGMP mediated vasodilatation [22]. Endotoxin enhances this vasodilatory response [100]. As cardiac filling pressures increase extra-vascular lung water (EVLW) increases, this is compounded by the increased vascular permeability characteristic of sepsis [101]. An increase in the central venous pressure (CVP) profoundly decreases microcirculatory flow and impairs renal function (see end-points of resuscitation below). The increased cardiac filling pressures consequent to large volume resuscitation increases the release of natriuretic peptides [102, 103]. Natriuretic peptides cleave membrane-bound proteoglycans and glycoproteins (most notably syndecan-1 and hyaluronic acid) off the endothelial glycocalyx (see Chap. 9) [104–106]. This profoundly increases endothelial permeability. In addition, increased natriuretic peptides inhibit the lymphatic propulsive motor activity reducing lymphatic drainage [107–109]. Increased natriuretic peptides therefore act to sequestrate fluid into the interstitium. Humans/mammals have evolved over thousands of years to deal with hypovolemia; hypervolemia is a rather recent and largely “iatrogenic” condition.
Emerging data suggests that less than 50 % of septic patients who present to the ER (and are fluid naive) will be fluid responsive! As reviewed in Chap. 9 only patients who are fluid responsive should be given fluids; fluids are likely to be harmful in non-responders. This suggests that patients’ fluid responsiveness status should be evaluated prior to fluid therapy. In addition, it is important to emphasize that the Frank-Starling curve is shifted downwards and to the right in sepsis, with septic patients showing a diminished response to fluid loading. Depressed left ventricular performance with a reduced response to fluid infusion in patients with sepsis and septic shock was demonstrated by Ognibene et al. in a study published in 1988 [110]. Using data obtained from a pulmonary artery catheter and radionuclide cineangiography they compared the change in left ventricular stroke work index ((LVSWI) in critically ill control subjects, patients with sepsis but without shock and patients with septic shock. The septic patients had both a reduced LVEF and increased end-diastolic volume index (EDVI) suggesting systolic LV dysfunction. However, the change in EDVI was less in the patients with sepsis and septic shock than the controls, suggesting an element of diastolic dysfunction. The LVSWI response after volume infusion (about 1,000 mL NS) was significantly less in patients with septic shock when compared with critically ill control subjects (p < 0.05). Patients in septic shock had a minimal increase in LVSWI in response to volume infusion (see Fig. 12.3).
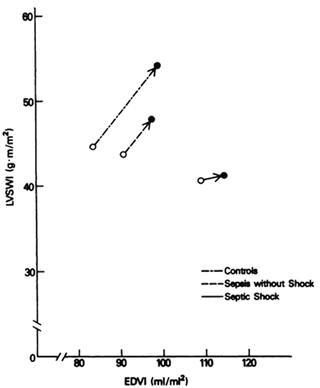
Fig. 12.3
Frank Starling relationship for control patient’s and those with sepsis and septic shock. Data points plotted represent the mean pre-volume and post-volume infusion values of EDVI and LVSWI for each group. Reproduced with permission from the American College of Chest Physicians [110]
Due to the endothelial injury, capillary leak and increased hydrostatic pressures less than 5 % of infused crystalloid remains intravascular within 3 h after infusion resulting in an further increase in EVLW and tissue edema [111]. Tissue edema impairs oxygen and metabolite diffusion, distorts tissue architecture, impedes capillary blood flow and lymphatic drainage and disturbs cell-cell interactions; these effects contribute to progressive organ dysfunction. These effects are pronounced in encapsulated organs, such as the liver and kidneys, which lack the capacity to accommodate additional volume without an increase in interstitial pressure, resulting in compromised organ blood flow [112]. This leads to AKI and hepatic congestion with cholestasis and impaired hepatic function. Increased EVLW (pulmonary edema) impairs gas exchange, reduces lung compliance and increases the work of breathing. Increased EVLW is a strong independent predictor of death [113–115]. Myocardial edema due to excess fluid administration compounds the myocardial dysfunction of sepsis [99]. Bowel edema results in malabsorption, ileus and bacterial translocation.
In patients with pneumonia, large volume fluid resuscitation results in severe pulmonary edema/ARDS (see Fig. 12.4). The chest radiographs presented in Fig. 12.4 represents a typical case of “fatal iatrogenic salt water drowning.” This patient was resuscitated according to the EGDT and the Surviving Sepsis Campaign bundles, in which fluids are administered until the CVP >12 mmHg [62, 72]. We know that patients’ with sepsis have damaged endothelium (especially at the site of infection, i.e. the lung) with increased permeability and that almost all the of administered crystalloid will leak into the tissues (i.e. the lung). This course of events is clearly evident from the CXR progression outlined in Fig. 12.3. Furthermore, patients with sepsis are primarily not dehydrated but suffering from vasoplegic shock with an increase in the non-stressed blood volume (veno-dilatation); it is therefore totally illogical (and stupid) to give large volumes of fluid to patients with sepsis.
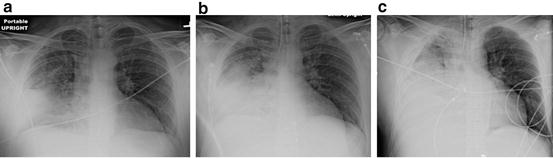
Fig. 12.4
(a–c) 44 year old male with Pneumococcal pneumonia. (a) Initial Chest radiograph (CXR) in emergency room; (b) CXR 4 h later after 4 L crystalloid (pt now intubated); (c) CXR 12 h after admission, after 9 L of crystalloid, central venous pressure (CVP) = 10 mmHg. Patient died 6 h later of refractory hypoxemia
The harmful effects of aggressive fluid resuscitation on the outcome of sepsis are supported by experimental studies as well as data accumulated from clinical trials [99, 116]. Multiple clinical studies have demonstrated an independent association between an increasingly positive fluid balance and increased mortality in patient with sepsis [7, 47, 114, 117–120]. In a secondary analysis of the Vasopressin in Septic Shock Trial (VASST), Boyd and colleagues demonstrated that a greater positive fluid balance at both 12 h and 4 days were independent predictors of death [121]. In a recent study Micek and colleagues demonstrated that a positive fluid balance at 8 days was the strongest independent predictor of hospital mortality [122]. In this study the 24 h fluid balance was 37.5 mL/kg (about 2.5 L) in the survivors compared to 55.3 mL/kg (3.9 L) in those who died. In patients with sepsis, Zang et al. demonstrated a strong correlation between the net fluid balance, the increase in BNP and death [103]. A number of studies have “paradoxically” demonstrated that a positive fluid balance was associated with an increased risk for AKI (see below; the danger of right atrial hypertension) [47]. Bouchard et al. demonstrated that in patients with AKI fluid overload was independently associated with mortality [120]. In the Fluid and Catheter Treatment Trial (FACTT) trial the conservative fluid strategy was associated with a trend towards a reduced requirement for renal replacement therapy [123]. There are no reported studies that have demonstrated that a liberal fluid strategy leads to improved patient outcomes [124]. The most compelling data that fluid loading in sepsis is harmful comes from “The Fluid Expansion as Supportive Therapy (FEAST)” study performed in 3,141 sub-Saharan children with severe sepsis [125]. In this randomized study aggressive fluid loading was associated with a significantly increased risk of death. Furthermore, there was no subgroup of patients that benefited from aggressive fluid resuscitation [126]. This study is frequently dismissed (by proponents of the Surviving Sepsis Campaign) using the argument that a study conducted in children cannot be extrapolated to adult patients [96]. This argument is absurd as studies performed in rats (who generally differ quite considerably from humans) are frequently extrapolated to humans yet the FEAST study was done in humans, albeit little ones.

In patients with sepsis almost 100 % of infused crystalloids will filter into the interstitium. This combined with decreased lymphatic drainage results in severe tissue edema and increased extravascular lung water.
THIS DOES NOT SEEM TO BE A GOOD THING or to MAKE ANY SENSE!
In contemporary sepsis studies between 1.5 and 4.0 L of fluid were given in the first 24 h [103, 122, 127]. This compares to 4.9 ± 2.9 and 13.4 ± 6.3 L at 6 and 72 h respectively in the intervention arm of the Early Goal Directed Therapy (EGDT) study [62]. In the Australasian Resuscitation of Sepsis Evaluation (ARISE) study which used the same entry criteria as the EGDT study, 2.2 ± 1.9 L of fluid were given in the first 6 h [127]. The hospital mortality was 23 % in the ARISE study compared to 30 % in the intervention arm of the EGDT study. In the VASST study optimal survival occurred with a positive fluid balance of approximately 3 L at 12 h [121]. In some patients, hypotension and tachycardia do resolve with limited fluid resuscitation. However, fluids alone will not reverse the hemodynamic instability of patients with more severe sepsis; in these patients’ fluids alone are likely to exacerbate the vasodilatory shock and increase the capillary leak, tissue edema and organ dysfunction. Based on these data we suggest limiting the initial fluid resuscitation to approximately 20 mL/kg. Furthermore, we recommend this fluid be given as 250–500 mL fluid challenges. Ideally, all patients should undergo assessment of fluid responsiveness prior to receiving any fluid. These recommendations differ fundamentally from those of the most recent Surviving Sepsis Campaign Guidelines which suggest “a minimum fluid challenge of 30 mL/kg” and that “greater amounts of fluid may be needed in some patients (Grade 1C).” [72] The EGDT protocol has no limit on the amount of fluid given; the target is a CVP >8 mmHg (12–15 mmHg if on a ventilator) regardless of the amount of fluid given [62]. This is likely to be a highly lethal approach as discussed below (see end-points of resuscitation). The fluid of choice in almost all septic patients is Ringers Lactate (this is reviewed in detail in Chap. 9). An infusion of 20 % albumin, to help restore the endothelial glycocalyx, may have a role in resuscitated patients with septic shock who have a low serum albumin (< 3 g/dL) (see Chap. 9) [128].
It is noteworthy that the amount of fluid given in the first 6 h and from 7 to 72 h was significantly less in the ProCESS EGDT patients than in the Rivers’ EGDT patients (See Table 12.2). As patients were resuscitated according to the same protocol this would appear to be odd. However, it should be noted that the mean CVP at 6 h was 13.8 ± 4.4 in the Rivers’ EGDT group. Assuming a normal distribution, 50 % of patients in the Rivers’ EGDT would have achieved CVPs greater than the mean value of 13.8 mmHg. Thus most patients in the Rivers’ EGDT study had CVPs outside the stated goal (>8–12 mmHg). It is possible that the enormous amount of fluid administered in the Rivers study partly accounted for the mortality difference between the EGDT arms of the Rivers’ and ProCESS studies. Furthermore, the use of vasopressors in the first 6 h of EGDT was significantly greater in ProCESS than in the Rivers’ study; this suggests the earlier use of pressors for blood pressure support in ProCESS (as we currently suggest; see below and Fig. 12.5)
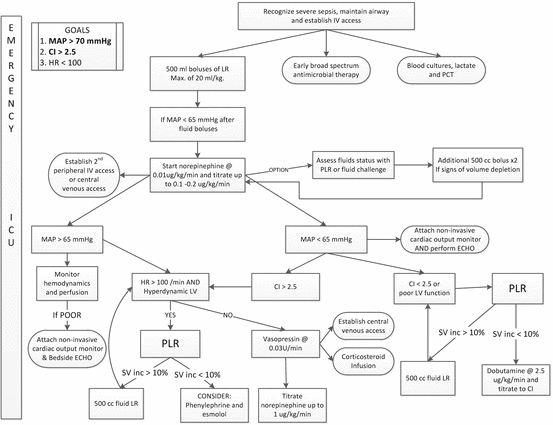
Table 12.2
Contrasting use of fluids and vasopressors (and mortality) in the EGDT arms of the Rivers’ and ProCESS studies
Study | Fluid | Fluid | Fluid | Vasopressors (%) 0–6 h | 60 day |
|---|---|---|---|---|---|
0–6 h (mL) | 7–72 h (mL) | 0–72 h (mL) | mortality (%) | ||
Rivers EGDT | 4,981 | 8,625 | 13,443 | 27.4 | 44.3 |
ProCESS EGDT | 2,805 | 4,428 | 7,220 | 54.9 | 21 |
Fig. 12.5
Suggested initial approach to the management of patients with severe sepsis and septic shock. MAP mean arterial pressure, LR Lactated Ringers solution, CI cardiac index, LV left ventricle, PLR passive leg raising, PCT procalcitonin
The optimal time to start a vasopressor agent in patients with sepsis has not been well studied. However, after receiving 20 mL/kg of crystalloid it seems unlikely that additional fluid boluses will increase the mean arterial pressure (MAP) in patients who remain hypotensive [95, 96]. We would therefore recommend the initiation of a vasopressor agent (norepinephrine) in patients who remain hypotensive (MAP <65 mmHg) after receiving 20 mL/kg of crystalloid solution. Additional fluid boluses (250 cm3) may be given once the “target” norepinephrine dose is achieved (about 0.1–0.2 μg/kg/min) and this is should be based on a dynamic assessment of volume responsiveness and ventricular function (see Fig. 12.5). We suggest using the passive leg raising manoeuvre (PLR) coupled with minimally invasive cardiac output monitoring to assess volume responsiveness (see Chap. 9).[129, 130] In cases of life-threatening hypotension (diastolic blood pressure <40 mmHg), treatment with vasopressors should be started concurrently with fluid administration [131]. As discussed in Chap. 9, Ringers Lactate is the fluid of choice. An albumin infusion should be considered in patients with a albumin concentration <3 g/dL.
Vasopressors and Inotropic Agents
A low MAP is a reliable predictor for the development of organ dysfunction. When the MAP falls below an organs autoregulatory threshold, organ blood flow decreases in an almost linear fashion [132]. Since the autoregulatory ranges of the heart, brain and kidney are above 60 mmHg [132], a MAP below this level will likely result in organ ischemia. An analysis of a large ICU database demonstrated that the risk of kidney injury and death increased sharply as the MAP fell below 60 mmHg [133]. Varpula and colleagues studied the hemodynamic variables associated with mortality in patients with septic shock [134]. These authors calculated the area under the curve (AUC) of various MAP thresholds over a 48 h time period. The highest AUC values were found for a MAP <65 mmHg (AUC 0.83, 95 % CI, 0.772–0.934). Due to the shift of the autoregulatory range (to the right) in patients with chronic hypertension a higher MAP may be required in these patients. The Assessment of Two Levels of Arterial Pressure on Survival in Patients With Septic Shock (SEPSISPAM) is a multicenter randomized controlled trial recently completed in France [135]. In this study patients with septic shock were randomized to achieve a target MAP of 65–70 or 80–85 mmHg. The primary outcome was 28 day mortality. Secondary outcomes included 90 day mortality and organ failures. A priori a secondary analysis was planned in patients with and without a history of hypertension. Overall there was no difference in either primary or secondary end-point between the two treatment groups. However the incidence of organ failures (particularly renal dysfunction) was higher in the sub-group of patients with chronic hypertension in the lower MAP group. Furthermore, much like the Varpula study, the time below the 65 mmHg (but not 80 mmHg) threshold was an independent predictor of death. It is important to recognize that the MAP in the 65–70 mmHg group exceeded the target threshold, with the average MAP being about 75 mmHg. Panwar et al. investigated the relationship between the mean perfusion pressure (MPP) deficit and the risk of AKI in 51 shocked patients [136]. The MPP deficit was calculated as the difference between the patients estimated basal MPP and the MPP achieved in the ICU. These authors demonstrated that the risk of AKI was related to the degree of the MPP deficit and the time spent with a >20 % MPP deficit. Based on these data we suggest targeting an initial MAP of 70 mmHg in patients with septic shock. In those patients with a history of chronic hypertension we would suggest targeting a slightly higher MAP (75–80 mmHg) [136].
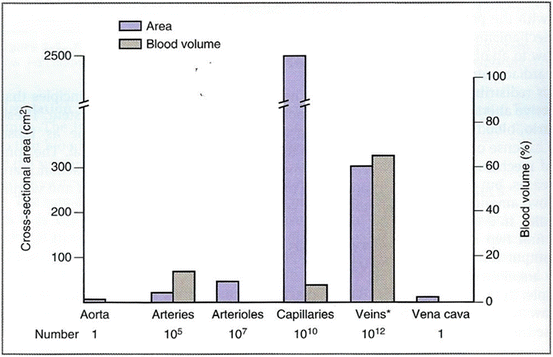
In patients with sepsis, norepinephrine increases blood pressure, as well as cardiac output, renal, splanchnic, cerebral and microvascular blood flow while minimally increasing heart rate [137–139]. Furthermore, while not widely appreciated, norepinephrine causes α-1 adrenergic receptor mediated veno-constriction; this effect increases the stressed blood volume and increases the mean systemic pressure (Pms) with a significant increase in venous return and cardiac output [140, 141]. It should be noted that approximately 60 % of the blood volume is in the veins; the unstressed blood volume increases with venodilation (see Fig. 12.6) Cardiac output (CO) equals venous return (VR). VR is related to the difference between Pms and CVP. Therefore, one can increase CO by increasing Pms and/or reducing the CVP, according to the following equation [142]:
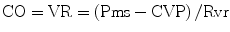 The early use of norepinephrine restores blood pressure and organ blood flow with a significant fluid sparing effect. Hamzaoui et al. have demonstrated that the early administration of norepinephrine largely reverses the hemodynamic abnormalities of severe vasodilatory shock [143]. Abid and colleagues demonstrated that the early use of norepinephrine in patients with septic shock was a strong predictor of survival [144]. In situations in which norepinephrine is not available epinephrine is a suitable alternative agent [145, 146]. In patients with septic shock dopamine is associated with an increased mortality when compared to norepinephrine and is best avoided [147, 148]. Similarly phenylephrine is not recommended, as in experimental models it decreases cardiac output as well as renal and splanchnic blood flow [149]. Furthermore, phenylephrine has not been well studied in patients with sepsis.
The early use of norepinephrine restores blood pressure and organ blood flow with a significant fluid sparing effect. Hamzaoui et al. have demonstrated that the early administration of norepinephrine largely reverses the hemodynamic abnormalities of severe vasodilatory shock [143]. Abid and colleagues demonstrated that the early use of norepinephrine in patients with septic shock was a strong predictor of survival [144]. In situations in which norepinephrine is not available epinephrine is a suitable alternative agent [145, 146]. In patients with septic shock dopamine is associated with an increased mortality when compared to norepinephrine and is best avoided [147, 148]. Similarly phenylephrine is not recommended, as in experimental models it decreases cardiac output as well as renal and splanchnic blood flow [149]. Furthermore, phenylephrine has not been well studied in patients with sepsis.
Fig. 12.6
Distribution of blood volume throughout the circulatory system
In patients who remain hypotensive or have evidence of inadequate organ perfusion despite fluid optimization and an adequate dose of norepinephrine (approximately 0.1–0.2 μg/kg/min) we recommend further hemodynamic assessment to exclude ventricular dysfunction. Global biventricular dysfunction has been reported in up to 60 % of patients with septic shock [26, 30]. Ventricular function is best assessed by bed-side echocardiography and confirmed by minimally invasive cardiac output monitoring. Dobutamine at a starting dose of 2.5 μg/kg min is recommended in patients with significant ventricular dysfunction (milrinone is an alternative agent) [30]. The dose of dobutamine should be titrated to hemodynamic response as determined by minimally invasive cardiac output monitoring [30, 150]. This approach differs from the EGDT protocol which suggests the use of an inotropic agent based on the CVP (> 8–12 mmHg) and a central venous oxygen saturation (ScvO2) of <70 % (without an evaluation of ventricular function or cardiac output) [62]. Bouferrache et al. demonstrated a poor agreement in the use of inotropic agents when treatment was guided by transesophageal echocardiography as compared to the EGDT protocol in patients presenting with septic shock [151].
Contrary to well established dogma, it appears to be reasonably safe to initiate norepinephrine via a peripheral venous catheter with few complications [152]. This is a remarkable observation as the greatest “resistance” to the early initiation of norepinephrine is the requirement for a central line. Placement of a second 18 or 20 gauge peripheral catheter will allow for the early initiation of norepinephrine. Once the dose reaches the threshold of about 0.2 μg/kg/min it is recommend to insert a central venous catheter at this time. Furthermore, central venous catheterization is recommend with the use of multiple vasopressor agents and in patients who require vaso-pressors for >48 h. Extravasation is a rare complication of well managed peripheral venous catheters; if this should occur the the steps outlined above are recommended.
Extravasation Injuries from Vasopressors
Prevention
Avoid the hand/wrist and ante-cubital fossa
Avoid using “Crappy” IV’s
A protocolized extremity check
Antidotes and a worksheet in the room with the patient.
10 mg of Phentolamine Mesylate can be added to each liter of solution containing norepinephrine. The pressor effect of norepinephrine is not affected.
Step I
If the patient is relying on the agent for their hemodynamics, switch the pressor to another IV or place an immediate central line.
Step II
Do not pull the cannula yet
Step III
Suck out as much fluid as you can
Step IV
Administer subcutaneous phentolamine mesylate (Regitine) using a 25 G or smaller needle. Phentolamine is available as 5 mg per 1 mL vials. Place in 9 mL of NS. A dose of 0.1–0.2 mg/kg (up to a maximum of 10 mg) should then be injected through the catheter and subcutaneously around the site. Administer as soon as the extravasation is detected, even if the area initially looks just a little white or OK. You should see near immediate effects; otherwise consider additional dose. Now pull the catheter.
Step V
Consult Plastics
Vasopressin reverses the “relative vasopressin deficiency” seen in patients with septic shock and increases adrenergic sensitivity [22, 153]. Terlipressin is an alternative (Terlipressin is not FDA approved in the US) [154, 155]. Vasopressin may be effective in raising blood pressure in patients with refractory hypotension however the optimal time to initiate this drug is not clear. The VASST trial randomized patients with septic shock to norepinephrine alone or norepinephrine plus vasopressin at 0.03 units/min [156]. By intention to treat analysis there was no difference in outcome between groups. However, an a priori defined subgroup analysis demonstrated that survival among patients receiving <0.2 μg/kg/min norepinephrine at the time of randomization was better with the addition of vasopressin than those receiving norepinephrine at a dose >0.2 μg/kg/min. We therefore suggest the addition of vasopressin at a dose of norepinephrine between 0.1 and 0.2 μg/kg/min. Thereafter the dose of norepinephrine should be titrated to achieve a MAP of at least 65 mmHg. It is important to emphasize that vasopressin is administered as a fixed dose of 0.03 units/min and should not be up-titrated. Our suggested treatment algorithm for the hemodynamic stabilization of patients with septic shock is provided in Fig. 12.5. It is however important to emphasize that “patients are not airplanes and doctors are not pilots”;[157] each patient is unique, with a unique response to invading pathogens and a unique response to treatment, therefore this algorithm must be dynamically adapted to each patient as his/her clinical course evolves.
Related posts:




Full access? Get Clinical Tree


