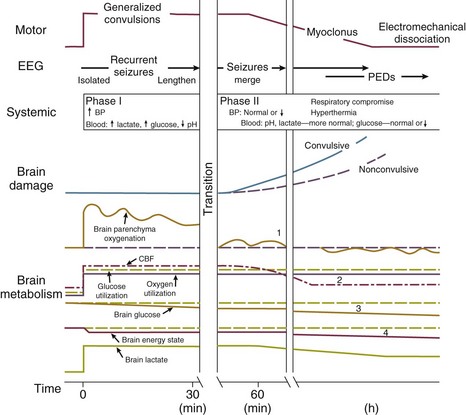
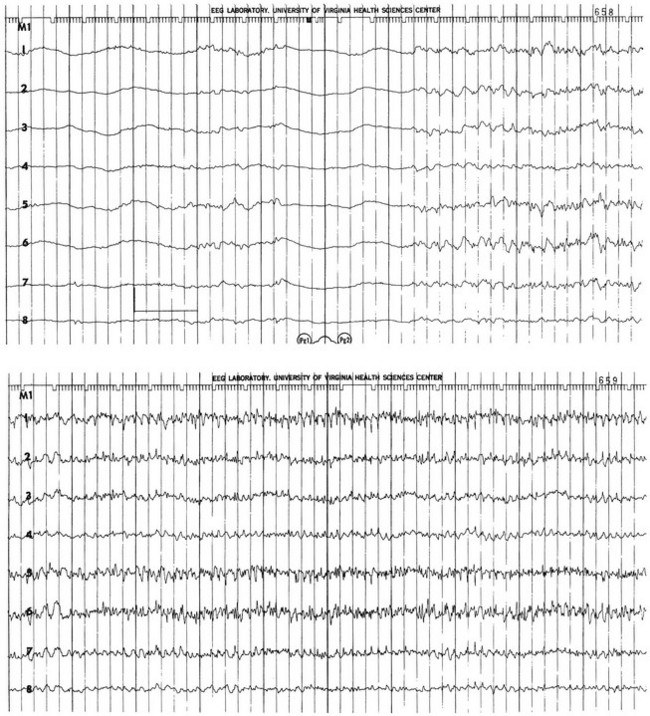
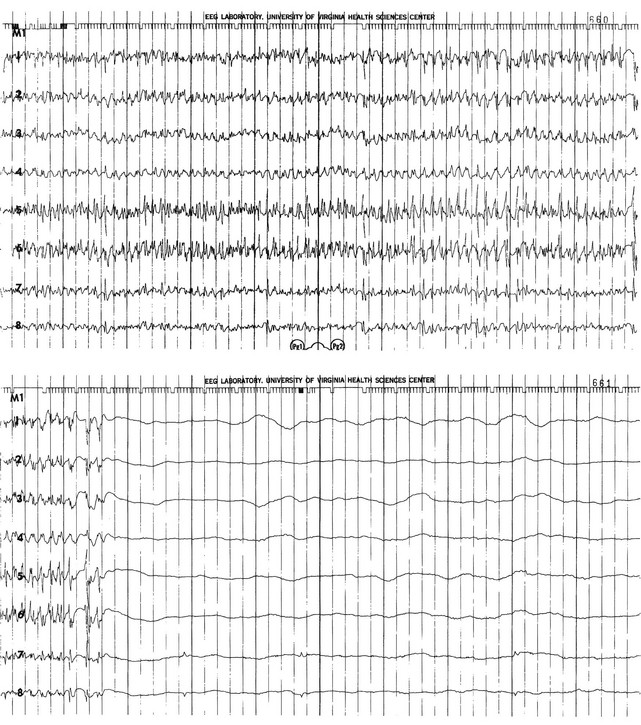
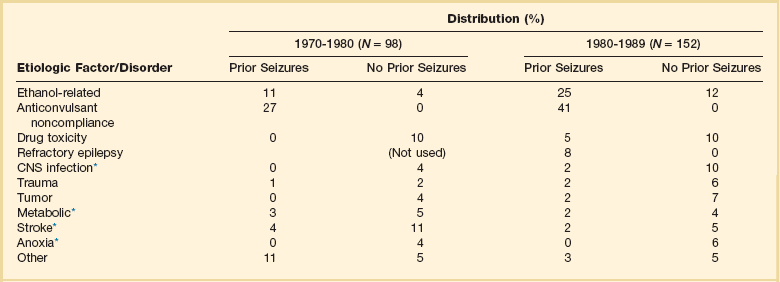
65 Seizures complicate the course of about 3% of adult patients admitted to intensive care units (ICUs) for non-neurologic conditions1 and occur more frequently in specialized neuroscience ICUs.2 The medical and economic impact of seizures in these patients confers a significance on these events out of proportion to their incidence. Seizures are often the first indication of a central nervous system (CNS) complication in these patients, making their rapid etiologic diagnosis mandatory. Furthermore, because epilepsy is a common disorder (affecting about 2% of the general population), patients with preexisting seizure disorders will occasionally require ICU admission for intercurrent conditions. The intensivist usually manages the initial treatment of these patients, so he or she must be familiar with the indications and risks of the potential therapies as they affect the already critically ill patient. In addition, the patient who develops status epilepticus (SE), whether already in the ICU or not, will often require the care of a critical care specialist in addition to a neurologist. The first recorded description of SE is by Gavasetti in 1586.3,4 Sir Thomas Willis described the complications of untreated SE in 1667: … as to what further belongs to the prognostication of the Disease, if it end not about the time of ripe age, neither can be driven away by the use of medicines, there happens yet a diverse event in several sick Patients, for it either ends immediately in Death, or is changed into some other Disease, to wit, the Palsie, stupidite, or melancholly, for the most part incurable. As to the former, whenas the fits are often repeated, and every time grow more cruell, the animal functions are quickly debilitated; and from thence, by the taint, by degrees brought on by the Spirits, and the Nerves serving the Praecordia, the vital function is by little and little enervated, till at length, the whole body languishing, and the pulse is loosned, and at length ceasing, at last the vital flame is extinguished.5 Attempts at treating SE in the nineteenth century included bromide,6 morphine,7 and ice applications. Barbiturates were introduced in 1912, followed by the identification and use of phenytoin in 1937; these were the first rational treatments for SE.8 Paraldehyde gained brief prominence in the next decades.9 The most recent major improvement is the use of benzodiazepines, pioneered by the French in the 1960s.10 Few data are available concerning the epidemiology of seizures in ICU patients. A 10-year retrospective study of all ICU patients at the Mayo Clinic reported approximately 7 patients with seizures per 1000 ICU admissions.11 In a 2-year prospective study of a medical ICU, we acquired approximately 35 patients with seizures per 1000 admissions.1 These analyses are not strictly comparable, because the patient populations and methods of detection differed. The incidence of seizures is probably higher in pediatric ICUs than in medical ICUs.12–14 Certain ICU patients appear to be at increased risk for seizures, but the degree of that increased risk has not been quantified. Patients with renal failure or with an altered blood-brain barrier who receive imipenem-cilastatin are an obvious example, but other patients receiving this antibiotic (or γ-aminobutyric acid [GABA] antagonists such as penicillin) occasionally seize. Cefepime has emerged as a cause of nonconvulsive seizures and SE, especially in patients with renal insufficiency.15 Transplant patients, especially those receiving cyclosporine, appear to have an increased risk for convulsions. Patients who rapidly become hypo-osmolar from any cause are also at risk. Nonketotic hyperglycemia patients have a high likelihood of partial seizures; this is a rare instance of a metabolic disorder producing focal neurologic syndromes.16 Less commonly, diabetic ketoacidosis may also produce partial seizures.17 The epidemiology of SE is somewhat better understood. Estimates of the incidence of generalized convulsive SE in the United States range from 50,000 cases/year18 to 250,000 cases/year.19 Some portion of this discrepancy may be due to differences in definitions. The larger estimate comes from the only population-based data available and may be more accurate. Similarly large variations occur in mortality rate estimates, from 1% to 2% in the former study to 22% in the latter. This disagreement stems, at least in part, from a conceptual discordance: the smaller number attempts to determine mortality rate that the authors directly attribute to SE, while the larger figure reflects the overall mortality rate for SE patients, in whom death was frequently a consequence of the cause of the underlying disease rather than SE itself. In the latter study, for example, anoxia was the cause of SE in adults with the highest mortality rate. In many of the reports surveyed in the earlier review, these patients were not included. Towne and colleagues demonstrated that 8% of an unselected series of comatose medical ICU patients had unsuspected nonconvulsive status epilepticus (NCSE).20 In septic ICU patients, Oddo and colleagues showed that about 30% have periodic epileptiform activity or NCSE when recorded for 24 hours or longer.21 Hospital-based series of SE patients are usually subject to considerable selection bias regarding cause. The data in Table 65.1, based upon 20 years of experience in San Francisco, are of great interest because almost all patients with SE in the city of San Francisco who began to seize outside the hospital are included.22–24 Table 65.1 Etiology of Status Epilepticus at San Francisco General Hospital *Conditions most likely to result in admission to intensive care unit (ICU). Between 6% and 12% of epilepsy patients present with SE,25 and about 20% of seizure patients will experience an episode of SE within 5 years of their first seizure.11 Numerous systems have evolved for the classification of seizures; the most frequently used today is that of the International League Against Epilepsy26 (Box 65.1). This schema allows classification based primarily on clinical criteria, without inferences about cause. Although a more recent proposal for terminology has been published, it is not yet widely accepted.27 It is important because of its predictive value for cause, prognosis, and treatment decisions in ICU patients. Simple partial seizures arise focally in the cerebral cortex, without taking over either the limbic system or subcortical nuclei. The patient remains aware of the environment during the ictus, and except for the seizure itself appears unchanged. Bilateral limbic system dysfunction results in a complex partial seizure; the patient’s awareness and ability to interact with the environment are diminished (but not always completely abolished). Automatisms are movements that the patient seems to make without being aware of them; typical automatisms include swallowing, masticatory movements, and fumbling with nearby items. Secondary generalization implies invasion of either the other hemisphere (with loss of consciousness) or, more commonly, subcortical structures, with the development of a generalized convulsion. Primary generalized seizures seem to arise from the entire cerebral cortex and the diencephalon at the same time; there are no visible focal phenomena. Consciousness is lost from the start of the seizure. True absence seizures are usually confined to childhood; they consist of the abrupt onset of a blank stare usually lasting 5 to 15 seconds, without lateralizing phenomena, from which the patient abruptly returns to normal. Atypical absence is usually seen in children who have the Lennox-Gastaut syndrome. Myoclonic seizures begin with brief, bilaterally synchronous jerks without an initial change in consciousness, followed by a generalized convulsion. They occur in several of the genetic epilepsies, but in the ICU are more commonly the consequences of anoxia or metabolic disturbances.28 Clonic seizures involve repetitive movements; they may be generalized (synchronous movements of all extremities and both sides of the face) or partial (e.g., one side of the face and the arm of the same side). Tonic seizures are episodes of tonic extension of the arms, legs, and trunk; they must be distinguished from decerebrate rigidity and from tetanic spasms.29 Tonic-clonic seizures begin with tonic extension, followed by a brief phase of rapid vibration of the extremities, evolving into bilaterally synchronous clonus, and concluding with a postictal phase in which incontinence is common and brief apnea is occasionally noted. They may be primarily generalized or, more commonly, occur as the manifestation of spread of a partial seizure. Only those seizures that are known to involve progression through the tonic and clonic stages should be called tonic-clonic. SE is classified by a somewhat similar system, with alterations to match the observable clinical phenomena (Box 65.2).30 Again, the ability to use clinical observation without inferences about cause is important. Generalized convulsive SE (GCSE) is the type most commonly encountered in ICUs, and poses the greatest risk to the patient. GCSE may be either primarily generalized, as in the intoxicated patient, or may represent secondary generalization, as in the patient with a brain abscess who develops GCSE. Tonic SE is usually seen in children or adolescents with a history of severe CNS dysfunction. Nonconvulsive SE (NCSE) in the ICU is most commonly the consequence of partially treated GCSE. Some authors use this as a general term for any SE involving altered consciousness without convulsive movements. Although conceptually useful, this blurs the distinctions among absence SE, partially treated GCSE, and complex partial SE (CPSE), which have different causes and treatments. Epilepsia partialis continua (EPC) is a special form of partial SE in which a small area of the body makes repetitive movements, sometimes for months or years following a CNS insult. The causes and effects of SE at the cellular, brain, and systemic levels are interrelated, but their individual analysis is useful for understanding them and their therapeutic implications. One must first understand the consequences of a single seizure and then contrast this information with the effects of prolonged or frequent seizures merging into SE. Longer durations of SE produce more profound alterations with an increasing likelihood of permanence, and of becoming refractory to treatment. Figure 65.1 illustrates the variety of processes involved in a single seizure and in the transition to SE.31 Many other important biophysical and biochemical alterations occur during and after SE. The intense neuronal activity activates immediate-early genes and produces heat shock proteins, providing strong indications of the deleterious effects of SE and insight into the mechanisms by which neurons protect themselves.32 Wasterlain’s group summarized the many mechanisms through which SE damages the nervous system.33 The electrical phenomena of SE at the whole brain level, as seen in the scalp electroencephalogram (EEG), reflect the seizure type that initiates SE (Fig. 65.2). Thus, absence SE begins with a generalized 3-Hz wave-and-spike EEG pattern. During the course of SE, there will usually be some slowing of this rhythm, but the wave-and-spike characteristic persists. In contrast, GCSE goes through the sequence of EEG changes outlined in Table 65.2. The initial high-frequency discharge becomes progressively less well formed over minutes; this pattern implies that neuronal activity is less synchronous. Whether this indicates that inhibitory systems are attempting to terminate SE, a progressive decay in the ability of synaptic mechanisms to maintain synchrony, or global deterioration in neuronal function remains to be determined. Table 65.2 Electrographic-Clinical Correlations in Generalized Convulsive Status Epilepticus *The clinical manifestations may vary considerably, depending on the underlying neuropathophysiologic process (and its anatomy), systemic diseases, and medications. In particular, stages of the electrographic progression may be sufficiently brief to be overlooked. Partially treating status epilepticus may dissociate the clinical and electrographic features. SE can produce cerebral edema, which follows ictal damage to the blood-brain barrier.
Seizures in the Critically Ill
History
Epidemiology
Nosology and Semiology
Pathophysiology
Stage
Typical Clinical Manifestations*
Electroencephalographic Features
1
Tonic-clonic convulsions; hypertension and hyperglycemia common
Discrete seizures with interictal slowing
2
Low- or medium-amplitude clonic activity, with rare convulsions
Waxing and waning of ictal discharges
3
Slight, but frequent, clonic activity, often confined to the eyes, face, or hands
Continuous ictal discharges
4
Rare episodes of slight clonic activity; hypotension and hypoglycemia become manifest
Continuous ictal discharges punctuated by flat periods
5
Coma without other manifestations of seizure activity
Periodic epileptiform discharges on a flat background
Related posts:

Full access? Get Clinical Tree


Anesthesia Key
Fastest Anesthesia & Intensive Care & Emergency Medicine Insight Engine