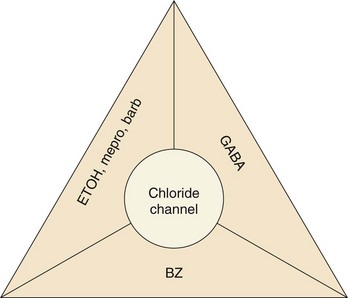
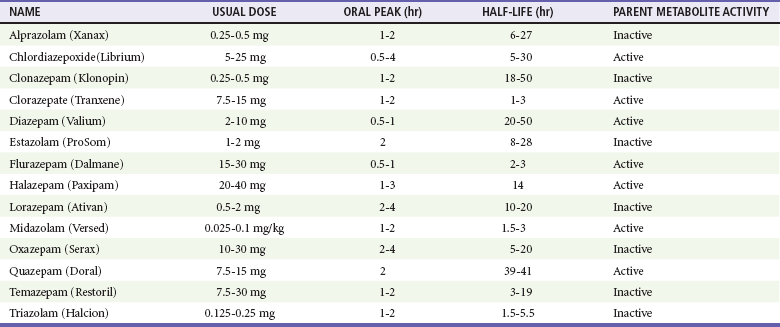
Chapter 165 Barbiturates are discussed in do-it-yourself suicide manuals and were implicated in the high-profile deaths of Marilyn Monroe, Jimi Hendrix, Abbie Hoffman, and Margaux Hemingway as well as in the mass suicide of 39 members of the Heaven’s Gate cult in 1997. Although barbiturates are still useful for seizure disorders, they rarely are prescribed as sedatives, with the availability of safer alternatives, such as benzodiazepines. Mortality from barbiturate poisoning declined from approximately 1500 deaths per year in the 1950s to only two fatalities in 2009.1 The GABAA receptor is a protein complex found on postsynaptic membranes in the CNS. Structurally, it consists of several distinct receptor sites surrounding a chloride ion (Cl−) channel (Fig. 165-1). GABA opens the chloride channel. The resulting flow of Cl− into the cell increases the negative resting potential, hyperpolarizing and stabilizing the membrane. There are separate receptor sites for barbiturates and for benzodiazepines and a third site that binds GABA, ethanol, and meprobamate. Although barbiturates and ethanol can directly increase Cl− conductance, benzodiazepines require the presence of GABA to affect Cl− flow, which may account for the relative safety of benzodiazepines in comparison with barbiturates. Barbiturates are classified according to their onset and duration of action (Box 165-1): ultra-short acting (onset immediate after intravenous dose, duration minutes), short acting (onset 10-15 minutes after oral dose, duration 6-8 hours), intermediate acting (onset 45-60 minutes, duration 10-12 hours), and long acting (onset 1 hour, duration 10-12 hours). Only long-acting preparations have anticonvulsant effects in doses that do not cause sedation. Short- and intermediate-acting preparations are almost completely metabolized to inactive metabolites in the liver, whereas 25% of a phenobarbital (long-acting) dose is excreted unchanged through the kidney. Because phenobarbital is a weak acid (pKa 7.2), alkalinization of the urine will increase the amount of drug present in ionized form, minimizing tubular reabsorption and increasing drug clearance. Short- and intermediate-acting barbiturates are not significantly affected by pH changes in this range. Gastric emptying by lavage is not indicated. For large overdoses, there is evidence that clearance of phenobarbital is markedly increased with multidose activated charcoal (MDAC).2 One dosage for MDAC is 25 g every 2 hours in an adult; the pediatric dose is 0.5 g/kg every 2 hours. If vomiting occurs, a smaller dose or antiemetics should be used. MDAC can also be administered slowly through a nasogastric tube. Contraindications to MDAC include an unprotected airway and gastrointestinal obstruction or perforation. Decreased peristalsis can result in constipation with MDAC and is a relative contraindication to MDAC.2 Although MDAC may shorten the duration of the intoxication, there is no evidence for improved outcome over supportive care, and supportive care without administration of activated charcoal is also an acceptable approach. Although alkalinization of the urine with sodium bicarbonate has been recommended in the past, a nonrandomized study suggested that MDAC alone is most effective at increasing the drug’s clearance.3 The authors of that study hypothesize that alkalinization may interfere with the ability of the drug to diffuse across intestinal mucosa from the blood into the gut. A recent comprehensive review concluded that there is no role for urine alkalinization in acute barbiturate poisoning.4 Hemodialysis is rarely needed but may increase clearance of phenobarbital in the presence of renal or cardiac failure, acid-base or electrolyte abnormalities, unstable cardiorespiratory status, or inadequate response to less invasive measures. Because phenobarbital is 40 to 60% protein bound, hemoperfusion was advocated over hemodialysis; however, newer high-efficiency dialyzers using high blood flow rates provide drug clearance greater than that achieved by hemoperfusion.5 Unfortunately, there are insufficient data to determine the true risk-benefit ratio of hemodialysis in acute barbiturate overdose.4 Benzodiazepines remain among the most widely prescribed class of drugs (Table 165-1) and are the most common prescription drugs used in suicide attempts. Fortunately, most benzodiazepine overdoses follow a relatively benign clinical course. Children make up 10% of benzodiazepine overdose cases. Benzodiazepines are rapidly absorbed orally. Intramuscular use of chlordiazepoxide and diazepam is limited by erratic absorption, but lorazepam and midazolam are predictably absorbed after intramuscular injection. After absorption, benzodiazepines distribute readily and rapidly penetrate the blood-brain barrier. In plasma, benzodiazepines are highly protein bound.6 All benzodiazepines are metabolized in the liver. Oxazepam, temazepam, and lorazepam are directly conjugated to an inactive, water-soluble glucuronide metabolite that is excreted by the kidney. Other benzodiazepines must first be converted by the hepatic cytochrome P450 system. Chlordiazepoxide, diazepam, flurazepam, and clorazepate are metabolized to active derivatives that are then slowly conjugated and excreted. The long elimination half-lives of these intermediates can cause accumulation in the body with repeated dosing. Triazolam, alprazolam, and midazolam are converted to hydroxylated intermediates that are active, but because they are so rapidly conjugated and excreted, they do not contribute significantly to the drug’s overall effect.6 Cytochrome P450 processes may be significantly impaired in elderly patients or those with liver disease, leading to prolonged elimination of some benzodiazepines. Coingestion of drugs that also undergo cytochrome P450 metabolism (e.g., cimetidine, ethanol) also prolongs the half-lives of these benzodiazepines, but the clinical significance of these interactions is unclear.7 CNS depression is common in patients with benzodiazepine poisoning and ranges from mild drowsiness to coma. Respiratory depression is due mainly to upper airway obstruction and increased upper airway resistance from loss of muscle tone rather than central apnea. Significant respiratory depression is rare but can be seen with large oral overdoses or during intravenous conscious sedation, particularly when the benzodiazepine is combined with an opioid such as fentanyl.7 Hypotension is uncommon. Other potential complications include aspiration pneumonia and pressure necrosis of skin and muscles. Prolonged or high-dose infusions of certain benzodiazepine preparations have been associated with the development of lactic acidosis. Metabolism of the propylene glycol diluent in diazepam and lorazepam intravenous solutions by alcohol dehydrogenase produces lactate, which can accumulate and cause acidosis severe enough to require intervention. Patients with renal or hepatic insufficiency are at increased risk for this complication.8 Any patient with altered mental status should have a blood glucose level rapidly determined. Qualitative immunoassays for benzodiazepines in urine are available but do not aid management decisions. Most of these tests detect only benzodiazepines that are metabolized to oxazepam glucuronide; therefore, clonazepam, lorazepam, midazolam, and alprazolam are not detected on many urine drug screens.9 Serum drug concentrations are not routinely available and do not correlate with clinical severity. A lack of alcohol odor or a negative breathalyzer or blood ethanol test result suggests benzodiazepine or another sedative as a possible cause. The benzodiazepine antagonist flumazenil should not be routinely administered to patients with coma of unknown origin or suspected benzodiazepine overdose, either for diagnostic or for therapeutic purposes.10 Any possibility of concomitant tricyclic overdose contraindicates flumazenil use. Initial stabilization, including endotracheal intubation, should not be delayed by the administration of an antidote. Most benzodiazepine overdoses can be managed expectantly. Activated charcoal is generally not beneficial in overdose.11 MDAC, hemodialysis, and whole-bowel irrigation are not effective in benzodiazepine overdose. Flumazenil, a nonspecific competitive antagonist of the benzodiazepine receptor, can reverse benzodiazepine-induced sedation after general anesthesia, procedural sedation, and confirmed benzodiazepine overdose, but it is not recommended for the routine reversal of sedative overdose in the ED. Although theoretic benefits of flumazenil use include cost savings and avoidance of procedures and tests such as endotracheal intubation and lumbar puncture, several studies have not been able to demonstrate an actual benefit.12 Seizures and cardiac dysrhythmias can occur after flumazenil administration, and fatalities have been reported. Flumazenil use can precipitate acute withdrawal in patients who are dependent on benzodiazepines. Similarly, this antidote is hazardous when it is given to patients who have coingested seizure-causing drugs (such as cocaine or a tricyclic antidepressant) because of loss of the benzodiazepine’s protective anticonvulsant properties. Coingestants that cause dysrhythmias, such as carbamazepine and chloral hydrate, may increase the likelihood of cardiac effects. Other risk factors are summarized in Box 165-2. One study found that 12% of patients receiving flumazenil after known pure or mixed benzodiazepine overdose actually had a contraindication to its use.13
Sedative Hypnotics
Barbiturates
Principles of Disease
Management
Gastrointestinal Decontamination and Enhanced Elimination
Benzodiazepines
Pharmacokinetics
Clinical Features
Diagnostic Strategies
Management
Antidote