Fig. 7.1
Metaxalone was approved as a muscle relaxant in the 1960s when two small studies suggested benefit in degree of low back spasm over the painful area and decreased pain interference; however, there has been a dearth of recent studies establishing either a mechanism of action or efficacy
Cyclobenzaprine
Cyclobenzaprine is one of the most commonly prescribed muscle relaxants, and while the exact mechanism by which it produces a muscle relaxant effect is not known, it may produce inhibition of serotonergic descending systems (Fig. 7.2) [15].
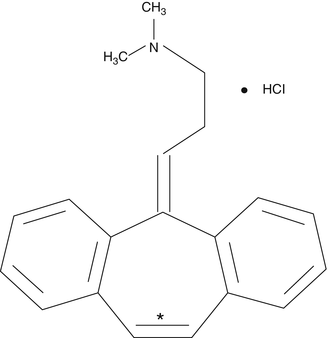
Fig. 7.2
Cyclobenzaprine is one of the most commonly prescribed muscle relaxants, and while the exact mechanism by which it produces a muscle relaxant effect is not known, it may produce inhibition of serotonergic descending systems
Cyclobenzaprine is chemically related to amitriptyline from which it differs by only one double bond. Cyclo-benzaprine metabolites also differ from amitriptyline metabolites by only one double bond. When doing forensic testing for the presence of these drugs and their metabolites, it may be necessary to use advanced techniques, such as high-performance liquid chromatography with ultraviolet detection or gas chromatography with nitrogen-phosphorus detection [16]. Laboratory technology involving high-performance liquid chromatography and tandem mass spectrometry is currently able to rapidly and quantitatively measure the following eight muscle relaxants in human blood: afloqualone, chlorphenesin carbamate, chlorzoxazone, dantrolene, eperisone, methocarbamol, pridinol, and tolperisone [17].
A meta-analysis of studies comparing cyclobenzaprine with placebo showed efficacy to be greatest on day 4 and then declining after the first week. NNT = 3, meaning three patients required treatment for one to show response [18]. In this now 10-year-old paper, a strong recommendation was made for comparing efficacy among active controls such as acetaminophen and NSAIDs which has since been done. A 2010 study shows efficacy for cyclobenzaprine 5 mg TID, but no benefit over an NSAID (ibuprofen 800 mg TID) during a 7-day treatment of acute cervical pain presenting at the emergency department of a large university hospital [19]. In this small study of 61 patients, although findings did not reach statistical significance, pain was more quickly relieved in patients receiving cyclobenzaprine, and the degree of pain intensity relief was greater for cyclobenzaprine compared to ibuprofen and was greatest with a combination of cyclobenzaprine and the NSAID. Cyclobenzaprine is commonly prescribed at a dose of 10 mg TID for muscle spasm with local pain and tenderness, is thought to increase range of motion, and is associated with a high incidence of side effects such as drowsiness and xerostomia. Interestingly, an industry-funded dose ranging study suggests 5 mg TID produces less side effect while maintaining efficacy [20].
Cylobenzaprine, like the related tricyclic antidepressants and also opioids, activates toll-like receptors (TLR) in spinal microglial cells [21]. Glial cell activation can have profound effects modulating pain and affect opioid-induced analgesia and tolerance. A mechanism by which tricyclic antidepressant class drugs including amitriptyline, imipramine, desipramine, cyclobenzaprine, carbamazepine, and oxcarbazepine can potentiate opioid analgesia has been demonstrated in mice [21]. These findings may explain how these drugs function as analgesics in chronic pain syndromes.
Carisoprodol
Regarding the non-tricyclic antidepressant muscle relaxants, one of the most controversial is carisoprodol (Fig. 7.3). Compared to placebo, it demonstrates efficacy for relief from acute muscle spasm and improved functional status at doses of 250 mg QID, although it is usually prescribed at 350 mg QID, a dose associated with a higher incidence of adverse effects [22]. Ralph et al. suggest carisoprodol would be a better drug if prescribed at the lower dose of 250 mg; however, the study was industry-sponsored, and authors disclosed they served on a speaker’s bureau for the product [22].
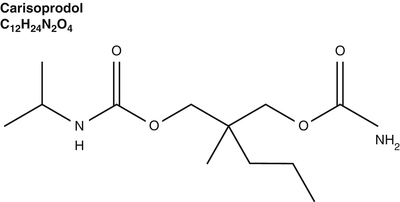
Fig. 7.3
Regarding the non-tricyclic antidepressant muscle relaxants, one of the most controversial is carisoprodol. Compared to placebo, it demonstrates efficacy for relief from acute muscle spasm and improved functional status at doses of 250 mg QID, although it is usually prescribed at 350 mg QID, a dose associated with a higher incidence of adverse effects
Carisoprodol is metabolized to meprobamate, an anxiolytic and hypnotic with known abuse potential, which also has a longer half-life. Either drug at a sufficient dose can produce mental impairment. An extensive database on nonalcoholic impaired drivers maintained in Norway includes extensive testing of mental function matched with forensic blood testing for drugs including carisoprodol and meprobamate. Impaired drivers admitted to consuming doses of carisoprodol greater than 700 mg and high carisoprodol levels correlated with impairment. Interestingly, Bramness et al. also reported that regular users of carisoprodol did not demonstrate high levels of meprobamate. The study was not designed to identify the mechanism though it was suggested that these patients had developed tolerance for the impairment caused by this active metabolite, while occasional users of carisoprodol who had not yet developed tolerance tended to have higher levels of meprobamate [23]. Metabolism of carisoprodol to meprobamate occurs via the CYP2C19 variant of cytochrome P450 in the liver. If there is variation of the cytochrome P450-CYP2C19 gene, it would be expected to affect meprobamate levels and subsequent side effects. For example, an individual with two CYP2C19 alleles may make more meprobamate and may have increased potential risk for impairment while driving [24].
An extreme case of withdrawal occurred in a patient taking a very high dose of carisoprodol, more than 17 g/day. Some might conclude that if such large doses could be tolerated, carisoprodol may actually have a high therapeutic index. In this case, withdrawal delirium occurred in a patient with back pain due to trauma who purchased large doses of carisoprodol over the internet when her health insurance lapsed. She was noted to be taking very high doses, up to fifty 350 mg tablets per day. She was not overly sedated and probably developed tolerance to the active metabolite, meprobamate. Seven days after deciding to stop, she lost orientation to person, place, and time and reported visual hallucinations, and postural and action tremors were noted on exam. Symptoms of delirium responded to treatment with 2 mg doses of lorazepam [25].
Concern for carisoprodol abuse since the Bramness study has led Norway to reclassify it as class-A (most restricted) led 39 of the United States to restrict its prescribing and led to a drive for the DEA to reclassify carisoprodol as a class-IV drug [26]. A case-control study was done in elderly patients identifying 8,164 cases and as many controls from a population of 1.5 million enrollees in a Medicare Advantage plan offered by a large HMO. Elderly patients receiving muscle relaxants were 1.4 times more likely to suffer a fracture injury, and the authors advised extreme caution be used prescribing muscle relaxants for older adults [27].
As our population ages, increased attention should be given to use and monitoring in elderly patients. Muscle relaxants are not recommended for patients over 65 years of age due to increased risk of injury due to side effects and should specifically to be avoided for elderly patients with bladder outflow obstruction and cognitive impairment [28].
However, while many reports as well as common wisdom advises against the use of muscle relaxants in the elderly, it has recently been suggested that skeletal muscle relaxants may be appropriate in this age group, especially if the patient does not have a high burden of disease and first-line medications were ineffective [29].
Baclofen
The muscle relaxants are a dichotomous group with indications for “skeletal muscle conditions” and for “spasticity” originating in the central nervous system, such as found in upper motor neuron disorders. Spasticity is an active muscle process whereby loss of central modulation causes increased excitability of the stretch reflex such that there is a velocity-sensitive response to limb manipulation [30]. Spasticity results from upper motor neuron pathology with abnormal stretch reflexes that may be the result of changed muscle structure, development of new spinal level collaterals, and/or failure to adequately regulate supraspinal pathways resulting in increased spinal reflex responses [31].
The traditional mainstay of treatment for upper motor neuron spasticity is baclofen, which has been used orally since the 1970s and, more recently, intrathecally (Fig. 7.4). To assess the possible survival advantage of intrathecal baclofen for cerebral palsy patients, 359 patients from Minnesota with intrathecal baclofen pumps were compared with 349 matched controls that were selected from 27,962 Californians with CP who did not have pumps. Interestingly, the survival for those with intrathecal baclofen was somewhat better than their well-matched controls [32].
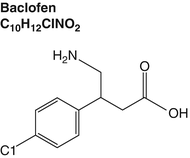
Fig. 7.4
The traditional mainstay of treatment for upper motor neuron spasticity is baclofen, which has been used orally since the 1970s and, more recently, intrathecally
Whereas benzodiazepines work at GABA-A receptors, increasing chloride ion currents causing cell hyperpolarization and thus inhibiting action potentials, baclofen activates the GABA-B receptor [33]. Designed to mimic GABA, baclofen is basically a GABA molecule with a chlorinated phenol moiety, hence its chemical name p-chlorophenyl-GABA. The only available prescription medicine that activates GABA-B receptors, baclofen has been the drug of choice for the treatment of tetanus, stiff man syndrome, cerebral palsy, and multiple sclerosis. In addition to treatment of spasticity, GABA-B receptor activation may also have a role in treatment of pain, depression and anxiety, drug addiction, and absence epilepsy, and GABA-B receptor antagonism may have a role in treating cognitive impairment [33].
Baclofen as a visceral pain reliever has been studied in sensitized visceral pain models where it appears to have a central site of action in the dorsal horn of the spinal cord at GABA-B receptors and, in a dose response fashion, attenuates both pain behavior and expression of FOS (a nociceptive marker). However, in the dose range that produced the analgesic effect, marked sedation was also observed [34].
In addition to the side effects of its use, in its withdrawal, baclofen may produce respiratory failure, unstable hemodynamics, seizures not responsive to usual treatment, and delirium. Interestingly, delirium is caused by both overdose and rapid withdrawal. If an intrathecal pump fails or needs to be removed due to infection, it is difficult using oral dosing to produce sufficient levels of baclofen in the CSF to prevent these catastrophic effects, and treatment with benzodiazepines, propofol, neuromuscular blocking agents, dantrolene, and tizanidine may be required in an ICU setting [35]. Baclofen and tizanidine withdrawal acutely produced extrapyramidal signs, delirium, and autonomic dysfunction that were eventually reversed when baclofen was restarted in a sufficient dose [36]. For a clear review of the differential diagnosis of baclofen withdrawal, the reader is referred to a recent case report with an excellent summary chart [37].
Other Muscle Relaxants
Of the muscle relaxants not available in the United States, one that should be mentioned is flupirtine (Fig. 7.5).
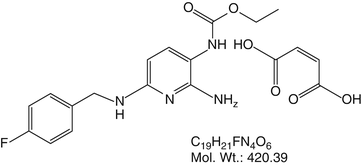
Fig. 7.5
Of the muscle relaxants not available in the United States, one that should be mentioned is flupirtine. Developed in Germany in the 1980s, flupirtine has been described as having many potential analgesic roles, and, equipotent to tramadol, it may also function as a muscle relaxant
Developed in Germany in the 1980s, flupirtine has been described as having many potential analgesic roles, and, equipotent to tramadol, it may also function as a muscle relaxant. Flupirtine activates Kv7 potassium channels, produces an M-current, and dampens hyperexcitable neurons [38]. The Kv7 potassium channel is activated by muscarine and is receiving a great deal of attention recently. There is speculation that further work could lead to new treatments for Alzheimer’s disease, seizure disorders, and chronic pain. The subtypes of Kv7 potassium channels regulate the potassium M-current activated by muscarine. Thus, muscarine (or other drugs acting at these sites) can lead to changes in potassium conductance with activation leading to hyperpolarization and blockade leading to increased neuronal activity. The M-current is a low-threshold, non-inactivating voltage-dependent potassium current at the Kv7 channel capable of limiting repetitive firing of neuronal action potentials [39]. Hyperexcitable states such as seizure disorders and chronic pain, including muscle pain and spasm, may respond to channel activators, while blockers at Kv7 channels might increase neuronal activation and provide a treatment of Alzheimer’s [39].
Used to treat painful contracture and spasticity, eperisone inhibits gamma-efferent firing in the spinal cord and produces local vasodilatation and rarely has adverse CNS effects (Fig. 7.6). It has good bioavailability, short onset time, and rapid elimination, making it suitable for initial treatment of acute low back pain [40].
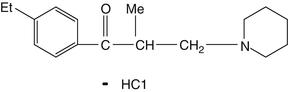
Fig. 7.6
Used to treat painful contracture and spasticity, eperisone inhibits gamma-efferent firing in the spinal cord and produces local vasodilatation and rarely has adverse CNS affects. It has good bioavailability, short onset time, and rapid elimination making it suitable for initial treatment of acute low back pain
While eperisone appears effective for treatment of muscle contracture and chronic low back pain, it is also touted to be free of sedative side effects [41]. Blood flow in low back muscles may increase with eperisone treatment over 4 weeks in comparison with placebo and active physical therapy protocols [42].
Related posts:




Full access? Get Clinical Tree


