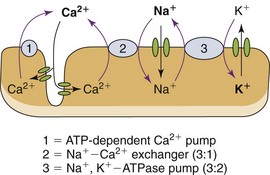
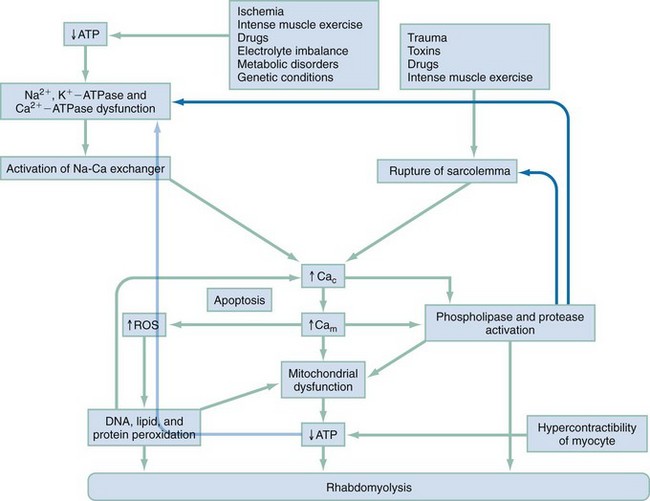
Chapter 127 A healthy 70-kg man has approximately 28 kg of muscle. Skeletal muscle is 80% water and 20% protein, accounting for about half of the total body protein stores, and is the largest organ in the human body.1 A large amount of adenosine triphosphate (ATP) is required for muscle function, even at rest. ATP generation by muscle accounts for 30% of the body’s oxygen consumption at rest and up to 85% at extremes of physical activity.2 Resting muscle uses fatty acids for ATP generation; muscle then draws on stored ATP for the first 8 seconds of activity, next using the phosphagen (creatine phosphate) stores for an additional 10 to 15 seconds; finally, muscle depends on anaerobic glycogen metabolism to lactate for enough ATP for an additional 30 to 40 seconds of activity. Aerobic ATP production provides the bulk of the energy needed for muscle activity but requires oxygen. Glucose, amino acids, and fatty acids are incorporated into the Krebs cycle to produce much larger quantities of ATP by energy-rich compounds, such as the reduced forms of nicotinamide adenine dinucleotide and flavin adenine dinucleotide. Under normal physiologic conditions, the concentration of free ionized calcium in the extracellular space is approximately 10,000 times greater than that in the intracellular space.3 The high concentration of free calcium in the extracellular pool compared with the intracellular compartment and the resulting large electrochemical force on Ca2+ are particularly convenient to its role as an intracellular regulator. Even minor changes in the permeability of the plasma membrane to calcium will produce significant fluctuations in the cytosolic concentration with potentially unfavorable consequences for the integrity of the cell.4,5 Several transmembrane proteins exist to regulate calcium homeostasis. The plasma membrane transmembrane proteins are the energy-consuming Ca2+ channels, the Na+-Ca2+ exchangers, and the Ca2+-ATPase pumps. The last removes Ca2+ from the intracellular space by transporting Ca2+ out of the cytosol into the extracellular space as well as into the sarcoplasmic reticulum. Plasma membrane Ca2+ channels bring Ca2+ into the cytosol when activated at the neuromuscular junction. Na+-Ca2+ exchangers are complicated, insofar that the direction of ion movement depends on the chemical and electrical gradients of each ion within the cell, which vary according to the contractile state of the myocyte3,6 (Fig. 127-1). Membrane damage from trauma and genetic or biochemical factors results in a massive influx of extracellular calcium into the cytoplasm driven by both electrical and chemical gradients.5 ATP depletion results in failure of cellular transport and increased permeability to sodium ions. Any event that increases cytosolic sodium concentrations, whether it is from increased membrane permeability and inward Na+ traffic or decreased ATP-dependent pump removal of intracellular Na+, results in increased Na+-Ca2+ ion exchanger function and increased cytosolic calcium concentrations. ATP depletion results in dysfunction of energy-dependent ion pumps, such as Na+,K+-ATPase and Ca2+-ATPase in the sarcolemma. Na+,K+-ATPase pump dysfunction leads to increased intracellular Na+, causing a temporary increase in Na+-Ca2+ exchanger function and resultant increase in intracellular Ca2+. The Na+-Ca2+ exchanger requires ATP, however, and continued activity of the Na+-Ca2+ exchanger further deprives the cell of ATP, leading to increased Ca2+-ATPase dysfunction and rising intracellular Ca2+ levels. The sarcoplasmic reticulum and the mitochondria are also equipped with these same energy-dependent transmembrane calcium transport mechanisms (Ca2+-ATPase) as well as an ATP-dependent Ca2+ uniporter, further exacerbating the cell’s inability to remove intracellular Ca2+ in the low-energy conditions put forth by rhabdomyolysis. Increased mitochondrial Ca2+ also leads to increased production of reactive oxygen species (ROS),7 which are a broad group of chemical substances that include free oxygen radicals (O2−, OH•) and powerful oxidants (hydrogen peroxide, nitric oxide) that lead to free radical production. Free radicals carry extra, unpaired electrons (e−) that have a strong tendency to pair off, causing oxidation by extraction of electrons from other chemical species. Under normal physiologic conditions, 2 to 5% of oxygen consumed by the mitochondria is transformed during electron transport to ROS that are neutralized by endogenous antioxidants such as glutathione. When the antioxidant capacity of the endogenous system is overwhelmed during times of intense, sustained physical activity or illness such that rhabdomyolysis results, a condition termed oxidative stress ensues. The resulting ROS-induced destruction of lipids and proteins injures the structure of membranes as well as DNA with mutations, leading to disruptions in cellular architecture and mitochondrial respiratory protein integrity. The cumulative effect exacerbates the inability to regulate intracellular Ca2+ homeostasis. Whereas ROS may have adaptive physiologic functions in exercising muscles, significant damage to muscle cells occurs when ROS are triggered by pathologic stimuli. Calcium-induced activation of proteases, phospholipases, and other proteolytic enzymes degrades the protein structure of myofibrils, the cytoskeleton, and membrane composition, leading to further cellular damage. Increased Ca2+ leads to sustained contraction of the myofibrils, resulting in severe ATP depletion in the initial stages while leading to myofibrillar breakdown as rhabdomyolysis progresses. Breakdown of the myofibrillar network hastens the disintegration of the myocyte, whereas high Ca2+ concentrations within the mitochondria arrest cellular respiration and block ATP production. Diminished ATP production after Ca2+-mediated activation of degradative cellular enzymes, with large metabolic energy requirements of their own, exacerbates the preexisting energy crisis within already ischemic cells.8 This process releases free fatty acids and lysophosphates that cause direct toxic damage to the sarcolemma, intracellular organelles, and plasma membrane carrier proteins, effecting increased Ca2+ entry into the cytosol. Microelectrode studies of intercostal myocytes show that intracellular calcium concentration rises to 1.27 µmol/L after induction of rhabdomyolysis by exhaustive physical exertion compared with 0.12 µmol/L in normal muscles.9 That is nearly an 11-fold increase in intracellular calcium concentration. In this study, dantrolene, a known inhibitor of Ca2+ release from the sarcoplasmic reticulum, was administered and led to an 83% reduction of cytosolic Ca2+ and an improvement in the clinical symptoms of muscle stiffness, rigidity, and pain, highlighting the essential role of increased cytosolic Ca2+ in the pathophysiologic process of rhabdomyolysis. In instances in which ischemia is the inciting event (e.g., crush syndrome, arterial thromboembolism) of rhabdomyolysis, myocyte destruction primarily takes place not during ischemia but rather during reperfusion10 (Fig. 127-2). Rhabdomyolysis is classified into four basic pathophysiologic processes: 1. Impairment of the muscle’s production or use of ATP at the cellular level. ATP concentrations within the cell fall; energy-dependent mechanisms falter, including, for instance, Na+,K+-ATPase pumps, leading to disruption of chemical gradients, sarcolemma and cell membrane compromise, and cell destruction. 2. Disruption in the delivery of oxygen, glucose, and other nutrients to skeletal muscle 3. Increases in metabolic demands beyond the ability of the organism to deliver oxygen and nutrients Box 127-1 lists the various causes of rhabdomyolysis. Rhabdomyolysis should be suspected in patients who present with altered mentation and risk factors for the development of rhabdomyolysis (e.g., intoxication, immobility, drug ingestion, electric shock, neuroleptic malignant syndrome). The presence of acute renal failure without any other obvious attributable cause also prompts consideration of rhabdomyolysis.11 The classic presentation of rhabdomyolysis includes localizing myalgias, muscle stiffness, cramping, swelling, tenderness, and tea-colored urine. The most frequently involved muscle groups are the thighs, calves, and lower back.12 Nonspecific constitutional symptoms include malaise, fever, nausea, and vomiting. Unfortunately, the majority of cases do not have characteristic physical signs; up to 50% of patients do not report myalgias or muscle weakness despite serologically proven rhabdomyolysis.12
Rhabdomyolysis
Principles of Disease
Pathophysiology
Etiology
Clinical Features
Rhabdomyolysis
Only gold members can continue reading. Log In or Register to continue





Full access? Get Clinical Tree


