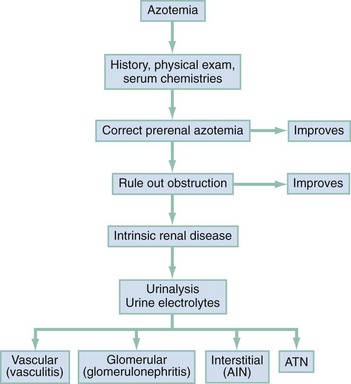
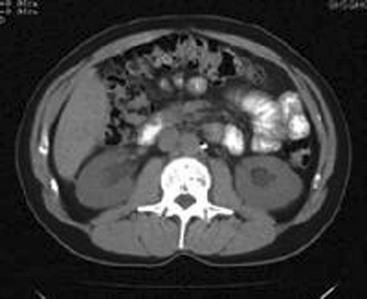
Chapter 97 The dipstick test for protein, which uses the color change of tetrabromophenol blue, can detect protein at concentrations of 10 to 15 mg/dL but does not yield reliably positive results until the concentration is greater than 30 mg/dL. Moreover, the relation between color intensity and protein concentration is only approximate. The dipstick reagent is three to five times more sensitive to albumin than to globulins and immunoglobulin light chains (e.g., Bence Jones protein)—an important limitation.1 False-positive results are caused by alkaline urine, hematuria, or prolonged immersion of the dipstick in the urine. False-negative results are seen with dilute urine. After dipstick testing of the urine has been completed, 10 mL of urine is placed in a conical test tube and spun at 2000 revolutions per minute for 5 minutes (higher speeds may break up casts). The supernatant is discarded. The sediment is resuspended in the residual urine, and a drop is placed on a slide and covered with a coverslip. Observations are recorded as the number of cells per high-power field. A level of two to three RBCs per high-power field in adult men or two to four RBCs per high-power field in adult women is commonly accepted as normal; in many studies a finding of five RBCs per high-power field is considered to represent the threshold of abnormality.2 Creatinine and Blood Urea Nitrogen.: The normal range for the serum creatinine level extends from 0.5 mg/dL in thin people to 1.5 mg/dL in muscular persons. Spurious elevations (up to 2 mg/dL) can be caused by acetoacetate (which cross-reacts with creatinine in commonly used assays) as well as by certain medications that either cross-react in the assay or reversibly inhibit tubular creatinine secretion despite a normal GFR (generally less than 0.5 mg/dL). Serum creatinine concentration is a function of the amount of creatinine entering the blood from muscle, its volume of distribution, and its rate of excretion. Because the first two are usually constant, changes in serum creatinine concentration generally reflect changes in GFR. The creatinine clearance is commonly estimated by the Cockcroft-Gault equation3: Urine Sodium and Fractional Excretion of Sodium.: Measurement of the urine sodium concentration provides information on the integrity of tubular reabsorptive function. Normally, urine sodium concentration parallels sodium intake. Low urine sodium concentration thus indicates not only intact reabsorptive function but also the presence of a stimulus to conserve sodium. The urine sodium concentration, as well as the fractional excretion of sodium (FENa), an additional measure of tubular sodium handling, helps distinguish between the two most common causes of AKI: prerenal azotemia and ATN (Table 97-1). Table 97-1 Typical Urinary Findings in Prerenal Azotemia and Acute Tubular Necrosis Urinary indices are most helpful in oliguric patients.4 In general, an oliguric patient with a urine sodium concentration below 20 mEq/L and an FENa below 1% should be considered to have prerenal azotemia, whereas urine sodium concentration above 40 mEq/L and FENa above 1% suggest ATN. Values in patients with prerenal azotemia overlap somewhat with those in patients with nonoliguric ATN, particularly if the renal injury is mild and some capability to retain sodium has been preserved. Thus, intermediate values for urine sodium concentration and FENa are of little help in differentiating between the two conditions. The administration of mannitol or a loop diuretic within the several hours preceding urine collection also may make interpretation of urine values difficult because the urinary sodium will tend to be higher and the urine less concentrated, causing the results in prerenal azotemia to resemble those in intrinsic renal failure (Box 97-1). Renal imaging is often helpful in evaluation of the patient with kidney dysfunction, particularly when obstruction is suspected. Contrast-enhanced computed tomography (CT) scanning provides an anatomic image of the urinary tract but does not provide an evaluation of renal function. The classic CT findings of obstruction are kidneys that are normal to large in size, nephrograms that become increasingly dense, and delayed opacification of dilated collecting systems. However, contrast-enhanced CT subjects the kidneys of an already azotemic patient to the risk of an additional potential insult from the contrast agent. Thus techniques such as ultrasonography and CT that do not involve contrast administration are much preferred in patients with preexisting renal insufficiency (Fig. 97-1). Figure 97-1 Computed tomography scan of bilateral hydronephrotic kidneys without intravenous contrast medium. Computed Tomography.: Noncontrast CT may be useful in evaluating some azotemic patients. Hydronephrosis can be recognized without the use of contrast material. Often, dilated ureters also can be seen without contrast enhancement, and the level of obstruction can be determined. Moreover, the cause of obstruction (e.g., bilateral stones, lymphoma, retroperitoneal hemorrhage, metastatic cancer, retroperitoneal fibrosis) often can be delineated as well. Occasionally, obstruction severe enough to result in renal failure may not cause detectable proximal dilation of the urinary tract. Bilateral ureteral obstruction produced by malignancy or retroperitoneal fibrosis is the most important cause of this nondilated obstructive uropathy. When noninvasive studies yield negative results, the diagnosis of obstruction can be made by retrograde pyelography or by antegrade pyelography performed via a percutaneous nephrostomy. Ultrasonography.: Ultrasonography allows accurate measurement of renal dimensions and is a safe and reasonably reliable method of excluding obstruction as a cause of AKI. The normal kidney shows an echo-free renal parenchyma surrounding the echogenic central urothelium of the renal pelvis and calices. The sonographic appearance of the kidney in obstruction is that of an enlarged central sonolucent area that spreads the normal central echo densities. A similar pattern may be produced by renal cysts, but without associated ureteral dilation. Dilation of the collecting system generally is apparent within 24 to 36 hours of the onset of obstruction, but obstruction may not be evident in patients who are evaluated early in the development of obstructive AKI. Microscopic hematuria often is discovered incidentally on routine urinalysis, but as little as 1 mL of blood in 1 L of urine can cause grossly appreciable hematuria. Although not invariably a sign of disease, the finding of hematuria calls for an effort to rule out any treatable underlying disorder. Both gross and microscopic hematuria are caused by similar disorders, but the amount of blood in the urine does not correlate with the severity or the seriousness of the underlying condition.2 The causes of hematuria can be divided into hematologic, renal, and postrenal; renal causes may be further classified as glomerular or nonglomerular (Box 97-2). Overall, the most common causes of nontraumatic hematuria, in roughly descending order of occurrence, are kidney stones, urinary tract infection (UTI), carcinoma of the kidney or bladder, urethritis, benign prostatic hypertrophy, and glomerulonephritis. The scope of the differential diagnosis can be narrowed by taking into account the patient’s age and sex and by distinguishing between upper and lower urinary tract sources. When gross hematuria is present, cystoscopy can determine whether blood is emerging from one or both ureteral orifices, thereby defining a source in the upper tract. Red cell casts indicate a renal source, as does associated proteinuria (excretion of more than 500 mg of albumin in 24 hours). In differentiating between proteinuria from renal parenchymal disease and that simply produced by admixture of urine with extravasated blood, a useful rule of thumb is that 1 mL of whole blood contains approximately 5 billion RBCs and approximately 50 mg of albumin. Other clues to the cause are obtained by careful questioning. Because glomerulonephritis or interstitial nephritis may be caused by a variety of bacterial, viral, and parasitic infections, a history of recent infection is important. Symptoms suggestive of a multisystem disorder (e.g., systemic lupus erythematosus) also should be sought, as should a history of human immunodeficiency virus infection.5 Because drugs may cause acute interstitial nephritis (AIN), papillary necrosis, or hemorrhagic cystitis, a complete medication history is elicited. When hematuria is associated with anticoagulant use, significant underlying disease can be identified in about one third of patients.6 The family history may provide a clue to the presence of polycystic or other familial kidney disease, sickle cell disease, or renal calculi. A history of recent strenuous exercise is important to identify; 15 to 20% of normal persons exhibit hematuria after strenuous exercise. The mechanism is unclear, but the hematuria resolves spontaneously within a few days. The role of urinary tract imaging studies in the immediate evaluation of hematuria also is limited. Visualization of the urinary tract generally is helpful only when the history suggests renal colic or other disorders of the upper urinary tract (e.g., polycystic kidney disease, tumor, obstruction). CT without contrast is the imaging modality of choice.7 Ultrasonography can be used to determine kidney size and shape and to detect renal masses or obstruction. Further imaging studies, if indicated, should be planned after urologic consultation. Patients with the nephrotic syndrome are at increased risk for thromboembolic events, including deep vein thrombosis of the lower extremity, renal vein thrombosis, and pulmonary embolism. The reason for this propensity appears to be a hypercoagulable state that is complex and incompletely understood.8 Hyperlipidemia is another typical feature of the nephrotic syndrome; the mechanism is thought to be related indirectly to hypoalbuminemia and decreased oncotic pressure or viscosity. The major clinical significance of the nephrotic syndrome, however, is that it indicates the presence of an underlying renal process or systemic disease affecting the glomerulus (Box 97-3). The hallmark of AKI (formerly known as acute renal failure [ARF]) is progressive azotemia, which commonly is accompanied by a wide range of other disturbances, depending on the severity and duration of renal dysfunction. These include metabolic derangements (e.g., metabolic acidosis and hyperkalemia), disturbances of body fluid balance (particularly volume overload), and a variety of effects on almost every organ system (Box 97-4). The causes of AKI may be divided into those that decrease renal blood flow (prerenal), produce a renal parenchymal insult (intrarenal), or obstruct urine flow (obstructive or postrenal). Identification of either a prerenal or a postrenal cause of AKI generally makes it possible to initiate specific corrective therapy; if these two broad categories of AKI can be excluded, an intrarenal cause is implicated. The renal parenchymal causes of AKI can be usefully subdivided into those primarily affecting the glomeruli, the intrarenal vasculature, or the renal interstitium. The term acute tubular necrosis denotes another broad category of intrinsic renal failure that cannot be attributed to specific glomerular, vascular, or interstitial causes (Fig. 97-2).4 Decreased renal perfusion that is sufficient to cause a decrease in the GFR results in azotemia. The possible causes can be grouped into entities causing intravascular volume depletion, volume redistribution, or decreased cardiac output (Box 97-5). Patients who have preexisting renal disease are particularly sensitive to the effects of diminished renal perfusion. Patients who have congestive heart failure (CHF) or cirrhosis form an important subset of those with prerenal azotemia. These patients often are salt-overloaded and water-overloaded, yet their effective intra-arterial volume is decreased. Administration of diuretics has the potential to decrease intravascular volume further, resulting in decreased glomerular filtration and prerenal azotemia. For some patients with advanced CHF or hepatic disease, a state of chronic stable prerenal azotemia may be the best achievable compromise between symptomatic volume overload and severe renal hypoperfusion.9 Glomerular perfusion also may be decreased in patients with normal intravascular volume and normal renal blood flow who take angiotensin-converting enzyme (ACE) inhibitors or, more commonly, prostaglandin inhibitors. All nonsteroidal anti-inflammatory drugs (NSAIDs), including aspirin, inhibit prostaglandin synthesis. Renal vasodilator prostaglandins are critical in maintaining glomerular perfusion in patients with conditions such as CHF, chronic renal insufficiency, and cirrhosis, in which elevated circulating levels of renin and angiotensin II decrease renal blood flow and GFR. In this setting, a decrease in the production of vasodilator prostaglandins may result in acute intrarenal hemodynamic changes and a reversible decrease in renal function.10 This phenomenon also is seen with the selective cyclooxygenase-2 inhibitor class of NSAIDs.11,12 Other risk factors include advanced age, diuretic use, renovascular disease, and diabetes. This entity is distinct from other renal complications of NSAIDs, including interstitial nephritis and papillary necrosis. Obstruction is an eminently reversible cause of AKI and should be considered in every patient with newly discovered azotemia or worsening renal function. Obstruction may occur at any level of the urinary tract but most commonly is produced by prostatic hypertrophy or by functional bladder neck obstruction (e.g., secondary to medication side effects or neurogenic bladder) (Box 97-6). Intrarenal obstruction may result from intratubular precipitation of uric acid crystals (e.g., with tumor lysis), oxalic acid (as in ethylene glycol ingestion), phosphates, myeloma proteins, methotrexate, sulfadiazine, acyclovir, or indinavir.13 Bilateral ureteral obstruction (or obstruction of the ureter of a solitary kidney) may be caused by retroperitoneal fibrosis, tumor, surgical misadventure, stones, or blood clots. A sudden deterioration in renal function in the setting of diabetes mellitus, analgesic nephropathy, or sickle cell disease should suggest papillary necrosis. Of the specific intrarenal disorders that cause AKI, glomerulonephritis, interstitial nephritis, and abnormalities of the intrarenal vasculature are amenable to specific therapy and are important to consider as possible causes. These entities are responsible for only 5 to 10% of cases of AKI in adult inpatients; most are caused by ATN. In adults in whom AKI develops outside the hospital, the incidence of glomerular, interstitial, and small-vessel disease is much greater. In children, these entities account for approximately one half of the cases of AKI (Box 97-7).4 Glomerular Disease.: Acute glomerulonephritis may represent a primary renal process or may be the manifestation of any of a wide range of other disease entities (see Box 97-7). Patients may have dark urine, hypertension, edema, or CHF (secondary to volume overload) or may be completely asymptomatic, in which case the diagnosis rests on an incidental finding on urinalysis. The hematuria associated with glomerular disease may be microscopic or gross and may be persistent or intermittent. Proteinuria, although often in the range of 500 mg/day to 3 g/day, not uncommonly is in the nephrotic range. The presence of hematuria, proteinuria, or red cell casts is highly suggestive of glomerulonephritis. In fact, red cell casts are essentially diagnostic of active glomerular disease, although occasionally they are seen with other types of renal disease. Conversely, the absence of red cell casts, proteinuria, and hematuria essentially excludes glomerulonephritis as the cause of AKI. Interstitial Disease.: AIN most commonly is precipitated by drug exposure or by infection.14 Drug-induced AIN is poorly understood, but the absence of a clear relationship to the dose and the recurrence of the syndrome on rechallenge with the offending agent suggests that an immunologic mechanism is responsible. The most commonly incriminated drugs are the penicillins, diuretics, and NSAIDs. AIN has been reported in association with bacterial, fungal, protozoan, and rickettsial infections. Patients with AIN classically have rash, fever, eosinophilia, and eosinophiluria, but it is common for one or more of these cardinal signs to be absent.15 Pyuria, gross or microscopic hematuria, and mild proteinuria are observed in some cases. A definite diagnosis sometimes can be made only on renal biopsy. Treatment of AIN is directed at removing the presumed cause; infections should be treated and offending drugs discontinued. Renal function generally returns to baseline over several weeks, although chronic renal failure has been reported to occur. Intrarenal Vascular Disease.: Vascular disease of the kidney can be classified according to the size of the vessel that is affected. Disorders such as renal arterial thrombosis or embolism, which affect large blood vessels, must be bilateral (or affect a single functioning kidney) to produce AKI. Whether to attribute such cases of AKI to prerenal or intrarenal vascular causes is a matter of semantics. The most common cause of thrombosis probably is trauma; thrombosis also may occur after angiography or may be secondary to aortic or renal arterial dissection. Renal atheroembolism is thought to occur commonly—at least on a microscopic level—after arteriography but is an uncommon cause of AKI. Similarly, patients with chronic atrial fibrillation or infective endocarditis may experience embolization of the kidney but rarely develop AKI as a result. Renal arterial embolism can cause acute renal infarction, generally manifested by sudden flank, back, chest, or upper abdominal pain. Urinary findings, including hematuria, are variable. Fever, nausea, and vomiting are not uncommon; in some cases, evidence of embolization to other vessels provides a useful clue. The diagnosis usually is made by renal flow scanning or arteriography. Surgical embolectomy has been reported to restore function when undertaken within several hours of occlusion, but significant return of function has been documented in patients operated on as long as 6 weeks after total occlusion. This outcome presumably is possible because collateral circulation has developed in association with a preexisting partial occlusion. Several diseases that affect the smaller intrarenal vessels can cause AKI (see Box 97-7). Patients whose disease is severe enough to cause ARF also generally are found to have hypertension, microangiopathic hemolytic anemia, and other systemic and organ-specific manifestations. Infection with Escherichia coli O157:H7 has emerged as a major cause of hemolytic uremic syndrome, an important cause of AKI in children.16 Malignant hypertension, although much less common since the advent of more effective antihypertensive therapy, has by no means disappeared. Patients with scleroderma (systemic sclerosis) may have “scleroderma renal crisis,”17,18 characterized by malignant hypertension and rapidly progressive renal failure. Whereas vasculitis associated with glomerular capillary inflammation typically causes gross or microscopic hematuria and formation of red cell casts, vascular involvement of the medium-size vessels, such as that produced by scleroderma, often spares the preglomerular vessels and tends not to produce an active urine sediment. Extrarenal manifestations (rash, fever, arthritis, pulmonary symptoms) are usually evident. For malignant hypertension, both as a separate entity and as a part of scleroderma renal crisis, appropriate treatment can produce a gratifying remission of AKI. Patients with malignant hypertension have been reported to recover renal function after aggressive antihypertensive therapy, with temporary maintenance with dialysis if necessary.19 In patients with scleroderma renal crisis, specific therapy with ACE inhibitors has been shown to result in improvement in renal function in a significant proportion of cases.18 The most common precipitant of ATN is renal ischemia occurring during surgery or after trauma and sepsis.20 The remainder of cases occur in the setting of medical illness, most commonly as a result of the administration of nephrotoxic aminoglycoside antibiotics or radiocontrast agents or in association with rhabdomyolysis. Multiple causes can be identified in some cases; in others a definitive cause is never established. ATN is common in postoperative patients, although not all cases can be attributed to intraoperative hypotension or hemorrhage. Concomitant sepsis, increased age, preexisting renal disease, and other comorbid conditions are associated with a worse outcome.20,21 Nephrotoxins constitute the other major cause of ATN. Among the most prominent of these are the endogenous pigments myoglobin and hemoglobin. Rhabdomyolysis and ARF resulting from crush injuries first received widespread attention after their description in survivors of the London blitz during World War II, but many other causes of pigment nephropathy have been reported (Box 97-8). Hypotension secondary to fluid loss into damaged muscle is thought to worsen the effects of myoglobinuria on the renal tubule, as does acidemia. Hemolysis, resulting in the release of hemoglobin into the circulation and hemoglobinuria, can cause ATN but usually only in the presence of coexisting dehydration, acidosis, or other causes of decreased renal perfusion. ATN may be associated with the hemolysis of as little as 100 mL of blood.
Renal Failure
Evaluation of Renal Function
Diagnostic Strategies
Protein
Microscopic Examination
Serum and Urine Chemical Analysis
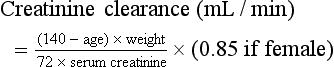
LABORATORY TEST FINDING
PRERENAL AZOTEMIA
ACUTE TUBULAR NECROSIS
Urinalysis
Normal or hyaline casts
Brown granular casts, cellular debris
Urine sodium concentration (mEq/L)
<20
>40
Fractional excretion of sodium (%)
<1
>1
Urine-to-plasma creatinine ratio
>40
<20
Renal Imaging
Hematuria and Proteinuria
Principles of Disease
Radiography and Ultrasonography
Proteinuria
Acute Kidney Injury
Principles of Disease
Postrenal (Obstructive) Acute Renal Failure
Intrinsic Acute Renal Failure
Acute Tubular Necrosis

Full access? Get Clinical Tree


Renal Failure