73 Pulmonary Edema
 Definition
Definition Pulmonary Fluid Homeostasis
Pulmonary Fluid Homeostasis
Pulmonary fluid homeostasis is dependent upon the equilibrium between forces that drive fluid into the alveolar space and counterforces responsible for its clearance—primarily lymphatics. One of the main regulatory forces for this fluid balance is the microvascular pressure in the alveolar capillaries, as presented by modification of the Starling’s equation (Figure 73-1, A), which describes the balance between the hydrostatic pressure gradient and the oncotic pressure gradient. While the oncotic or osmolarity gradient is dependent mainly on protein concentrations, the pulmonary capillary hydrostatic pressure is dependent on pulmonary flow and resistance (see Figure 73-1, B). Pulmonary capillary pressure is regulated at the precapillary level by the arteriolar vasomotor tone, which determines the transmission of flow and pressures from the pulmonary artery to the capillary bed. By contrast, venous capillaries lack this protective mechanism, allowing unprotected transmission of elevated left ventricular pressure to the pulmonary capillary bed and excessive fluid accumulation.1

Protective mechanisms against fluid accumulation in the alveolar and interstitial space include both passive elements, such as the tight junctions between the alveolar epithelium, and active reabsorption of fluid from the airspace using Na+ and Cl− channels.2 The primary sites of sodium and chloride reabsorption are the epithelial ion channels located on the apical membrane of alveolar epithelial cells (both type I and II) and the distal airway epithelial cells. Water will follow the osmotic gradient created by the reabsorption of Na+ and Cl−, preventing edema formation. Pulmonary edema will occur when this delicate balance is overwhelmed by one of three pathologic processes: impaired clearance mechanisms, increased hydrostatic pressures resulting in excessive pressure gradients, or increased permeability of the capillary alveolar barrier. When the main cause is related to increased pulmonary venous pressure, pulmonary edema is said to be cardiogenic in origin. In contrast, when other factors such as increased permeability prevail, the term noncardiogenic pulmonary edema is used. The interstitial fluid content in each etiology is different, owing to the underlying pathophysiology. Increased pulmonary venous pressures causing cardiogenic pulmonary edema will yield fluid with low protein content. Increased permeability of microvascular epithelium in noncardiogenic pulmonary edema will result in fluid with relatively high protein content.
 Diagnosis and Assessment
Diagnosis and Assessment
History and Presenting Symptoms
The presenting signs and symptoms of pulmonary edema are dyspnea, tachypnea, and respiratory distress. Alveolar flooding can lead to cough and expectoration of frothy edema fluid. The history should focus on cardiogenic and noncardiogenic mechanisms contributing to pulmonary edema and elicit precipitating factors that might have led to edema formation. Common causes for cardiogenic pulmonary edema include ischemia, exacerbation of systolic or diastolic dysfunction (ischemia, infarct, or myopathic processes), severe valvular disease, or arrhythmias. A history of paroxysmal nocturnal dyspnea or progressive orthopnea usually indicates cardiogenic origin for pulmonary edema. However, silent ischemia may also present as pulmonary edema, with a paucity of clues provided by the history.2 Noncardiogenic pulmonary edema is usually preceded by specific predisposing clinical situations such as pneumonia, sepsis, multiple blood transfusions, or intravenous (IV) illicit drug usage.
Physical Examination
Physical findings on lung examination are quite similar for cardiogenic and noncardiogenic pulmonary edema. The patient is usually tachypneic, pale, and diaphoretic with wet inspiratory rales/crackles heard over both lung fields, and most notably the bases. Patients with cardiogenic causes may present with an S3 “gallop” on cardiac auscultation, indicating elevated left-ventricular diastolic pressures—a sign with high specificity (90%-97%) but low sensitivity (9%-51%).2 Stenotic or regurgitant valvular murmurs on auscultation may indicate a cardiac cause but are not always related to the primary cause of the edema. Peripheral edema, which may be a sign for coexisting right heart failure, is neither sensitive nor specific for a cardiogenic origin of pulmonary edema. Most patients with cardiogenic causes for pulmonary edema will have cold, clammy skin, but some patients with noncardiogenic causes will present with warm skin, indicating decreased peripheral resistance.
Auxiliary Tests
Plain chest radiography has been reported to be more sensitive than clinical examination3 for pulmonary edema, which makes it one of the cornerstones for this diagnosis. The first finding that indicates interstitial edema are “Kerley B” lines. These are 3- to 6-mm-long lines perpendicular to the pleural surface, usually at the bases (Figure 73-2). Another sign of interstitial edema is peribronchial cuffing resulting from edematous thickening of the bronchial wall. Redistribution of blood to the upper fields of the lungs results in upper-lobe blood vessel distension. When fluid eventually leaks to the alveoli, bilateral and diffuse opacities are seen, usually sparing the apices and extreme lung bases, causing a central “butterfly” distribution. As the process progresses, opacities may coalesce to produce a general “white-out” of the lungs.4 Chest radiographs may aid in distinguishing between cardiogenic and noncardiogenic etiologies for pulmonary edema. In one study, it was demonstrated that in 50% of patients with cardiogenic edema there was upper-lobe blood diversion, whereas in patients with increased permeability edema due to acute respiratory distress syndrome (ARDS), only 10% showed this inverted pattern. Normal or “balanced” patterns were more commonly seen in ARDS. A peripheral distribution of edema was absent in patients with cardiogenic edema but was the most common pattern seen in patients with ARDS5 (Figure 73-3). Unfortunately, about one out of five patients admitted for acute decompensated heart failure had no signs of congestion on chest radiograph6—a fact that emphasizes the importance of a holistic, integrative approach to the diagnosis of pulmonary edema.
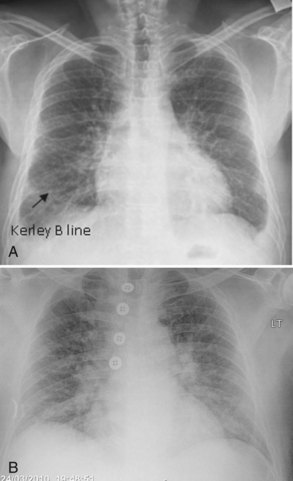
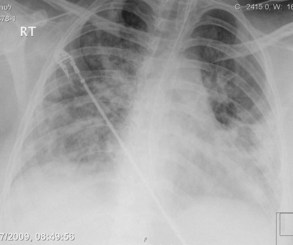
Figure 73-3 Chest x-ray images in ARDS. Diffuse bilateral opacities with involvement of peripheral lung fields.
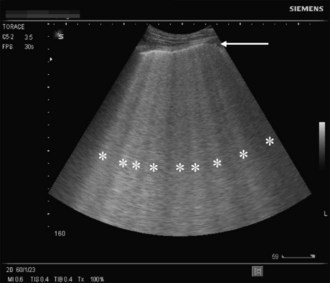
A novel approach utilizes ultrasound as a bedside tool to for the diagnosis of dyspnea and differentiation between pulmonary edema and other major dyspnea-causing diseases such as chronic obstructive pulmonary disease (COPD). Pulmonary edema induces abundant sonographic artifacts caused by interactions of water and air called B-lines or comet tails by some authors (Figure 73-4); these findings are usually not seen in other pulmonary diseases.7 Electrocardiograms are useful in diagnosing active myocardial ischemia or to provide other clues regarding organic cardiac disease leading the pulmonary congestion.
Biomarkers
In recent years, a variety of biomarkers have been used to enhance the diagnostic accuracy of cardiogenic pulmonary edema. Brain natriuretic peptide (BNP) and N-terminal proBNP (NT-proBNP) are both secreted from the ventricles and correlate with the left ventricular (LV) end-diastolic pressure; however, heart failure with preserved LV function usually results in much lower BNP levels than heart failure with impaired LV systolic function. These biomarkers can be used for several indications in the intensive care unit (ICU). Among others, it may aid in differentiating between cardiogenic pulmonary edema and acute lung injury (ALI), monitor volume load in septic patients, and differentiate between septic and cardiogenic shock. Several conflicting reports have addressed the use of BNP/NT-proBNP for the differentiation between ALI and cardiogenic pulmonary edema.8 Different cutoffs were used in the different trials, yielding various ranges of specificities and sensitivities for each diagnosis. It should be emphasized that these conflicting results arise in part from the fact that BNP increases with elevated right ventricular end-diastolic pressures and hypoxia, which are common properties of any severe lung disease. Based on currently available data, low levels of natriuretic peptide (BNP <100 pg/mL or NT-proBNP <250 pg/mL) may be used to exclude elevated cardiac filling pressures in patients presenting with respiratory failure with signs of pulmonary edema, whereas extremely elevated levels of these markers (BNP > 500 pg/mL and NT-proBNP > 1000 pg/mL) in the absence of signs and symptoms of septic shock will support a cardiogenic origin for pulmonary edema. The “gray zone” values between these extremes will necessitate further workup.8 Cardiac troponin (cTn) I or T measurement are highly sensitive for myocardial injury, which can aid in the diagnosis of cardiac origin of pulmonary edema, but in the setting of critical illness, various nonischemic conditions (sepsis, stroke, pulmonary embolism, acute renal failure, etc.) can also induce elevation of cTn and should be excluded before concluding that the cTn elevation is “ischemic.”
Echocardiography
Transthoracic echocardiography (TTE) is used to assist with the diagnosis of myocardial, valvular, and structural pathologies that contribute to pulmonary edema and thus should be performed in any patient presenting with pulmonary edema. While severe valvular stenosis or regurgitation is readily visible on echocardiographic exam, evaluation of cardiac function is more challenging. Decreased myocardial function in patients presenting with pulmonary edema can be due either to past myocardial ischemia/infarction or a current, ongoing ischemic event complicated by pulmonary edema. Furthermore, depressed myocardial function is often seen in other conditions associated with critical illness, such as sepsis. On the other hand, preserved systolic LV function cannot exclude a cardiac origin of pulmonary edema, since patients can present with heart failure and preserved LV systolic function (formerly diastolic dysfunction), thus necessitating further evaluation. Echo-Doppler can also provide semiquantitative measurements of ventricular filling pressures, cardiac output, stroke volume, and pulmonary artery pressures. TEE is used to enhance and refine evaluation of structural and valvular pathologies such as native or prosthetic valve dysfunction, cardiac origins of embolism, infective endocarditis, and congenital diseases.9 In one study performed in the ICU, TEE led to a significant change in management in 32% of cases,10 emphasizing its diagnostic value.
Hemodynamic Assessment
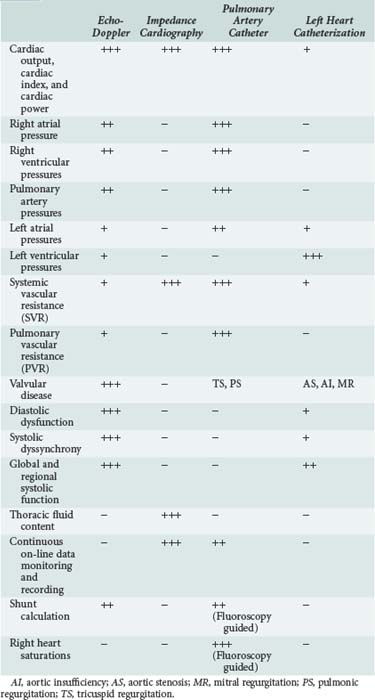
Pulmonary edema is a medical emergency and requires immediate medical therapy to alleviate symptoms. Dyspnea is the cardinal symptom of pulmonary edema and can be assessed subjectively (by analog scales of dyspnea severity) and objectively (by oxygen saturation, respiratory rate, alveolar-arterial difference, and acidemia). In most cases, the diagnosis, treatment, and monitoring of the patient with pulmonary edema is self-evident, but hemodynamic monitoring, either invasive or noninvasive, should be considered in selected patients. Since there are several techniques for hemodynamic assessment, the benefits and limitations of each technique should be considered prior to usage (Table 73-1).
Echo-Doppler
Pulmonary Artery Catheterization and Other Invasive Modalities
Insertion of a pulmonary artery catheter permits measurement of the pulmonary capillary wedge pressure (PCWP), a method first described in 1970 by Swan and Ganz12 and still considered to be the “gold standard” for diagnosis of pulmonary edema resulting from elevated LV diastolic filling pressures. Current monitoring systems that include cardiac output and systemic vascular resistance (SVR) calculators add further information and help distinguish cardiogenic pulmonary edema (high PCWP and high SVR) from noncardiac (low PCWP ± low/normal SVR). A wedge pressure of more than 18 mm Hg is indicative of elevated filling pressures of the left ventricle and usually indicates a cardiogenic origin of pulmonary edema. In addition to its utility in diagnosis, PCWP allows continuous monitoring of the LV filling pressure during treatment, facilitating the administration of appropriate therapy to alleviate pulmonary edema. It is recommended that pulmonary artery catheterization (PAC) be used in patients in whom a diagnostic dilemma exists, when echo-Doppler measurements are difficult to obtain, or in hemodynamically unstable patients not responding to conventional therapy.13
The clinical value and safety of PAC as a tool for hemodynamic assessment has been a subject of considerable debate. Gore14 and Connors15 demonstrated a neutral to negative effect of PAC on patient outcome. Meta-analyses assessing the effects of PAC on morbidity16 and mortality17 in clinical trials showed that mortality was unaffected, but morbidity was increased with the use of a PAC. There may be methodological issues in some of these studies; nevertheless, these publications resulted in a call for a moratorium18 on PAC. In 1997, a consensus conference19 attempted to reassess indications for PAC. Conditions that could be considered to benefit from PAC included myocardial infarction complicated by hypotension, shock, or mechanical complications, assessing and managing acute and chronic heart failure, and pulmonary hypertension. The ESCAPE study20 enrolled patients with established heart failure who did not require PAC for their diagnosis or management. The results of the study have demonstrated no benefit of right heart catheterization in the study’s primary endpoint (i.e., days alive out of hospital during the 6 months after randomization). Although the PAC is an invaluable tool for diagnostic, therapeutic, and prognostic assessment of PE, it should be used selectively by well-trained teams to address pertinent diagnostic and management issues.
Impedance Cardiography
Both thoracic impedance21 and total body impedance22,23 can accurately measure continuous CO and CI. ICG-derived COs appear to be less variable and more reproducible than CO measured by other techniques.24 Some bioimpedance systems, however, do not provide accurate CO values25 when compared to the gold standard of thermodilution.26 However, bioimpedance devices that can measure CO reliably can serve as tools for assessing pump performance by providing noninvasive measurements of CP and cardiac power index (CPI). None of these systems provide assessment of right side pressures or pulmonary vascular resistance (PVR).
Beat-to-Beat Pulse Contour
Semi-invasive techniques utilizing beat-to-beat pulse-contour analysis are becoming available for continuous cardiac output monitoring. At present, there are conflicting reports regarding the usefulness of these techniques compared to invasively measured PCWP in assessing the presence of pulmonary edema in different ICU populations,27–30 and no report has addressed the utility of pulse-contour analysis in differentiating between cardiac and noncardiac etiologies of pulmonary edema.
 Noncardiogenic Pulmonary Edema
Noncardiogenic Pulmonary Edema
A variety of etiologies may lead to noncardiogenic pulmonary edema (Table 73-2), with a final common pathway of fluid accumulation in the lung interstitium due to either increased permeability of capillaries or decreased fluid clearance mechanisms without evidence of elevation of LV end-diastolic pressure.
TABLE 73-2 Etiology of Noncardiogenic Pulmonary Edema
| Increased Capillary Permeability and Reduced Fluid Clearance |
| Toxins and Drugs |
| Alveolar-Capillary Pressure Imbalance |
| Perioperative pulmonary edema |
| Elevated Capillary Pressure (Fluid Shift/Excessive Fluid Transfusion) |
| Hypoxia Related |
| High-altitude pulmonary edema |
| Rapid Change in Intrathoracic Pressure |
Perioperative Pulmonary Edema
Perioperative pulmonary edema can result from a wide variety of etiologies including volume overload, negative pressure pulmonary edema (resulting from exaggerated negative intrathoracic pressure generated by an inspiratory effort against a closed glottis), and transfusions. In one large trial including 8159 patients undergoing major outpatient surgical procedures, an incidence of 7.6% of postoperative pulmonary edema was noted with approximately 12% mortality; of note, prior reports had reported lower rates of pulmonary edema and mortality.31,32 Excessive fluid administration during the postoperative period was associated with increased mortality, especially in patients without other comorbidities. Fluid overload during and after surgery can be attributed to exaggerated treatment for hypotension related to anesthesia, excessive blood loss, fluid shifts during surgery (“third spacing”), and postoperative fever. The relatively common incidence of pulmonary edema after surgery leads to a recommendation for close monitoring of fluid balance in the perioperative period, with special emphasis on monitoring patients at risk for developing pulmonary edema because of preexistent medical problems, including cardiac disease.
High-Altitude Pulmonary Edema
High-altitude pulmonary edema (HAPE) is the abnormal accumulation of edema involving the interstitial and alveolar spaces; it is due to a breakdown in the pulmonary blood-gas barrier. This is triggered by hypobaric hypoxia and rapid ascent to altitudes above 2500 m. Such hypoxia triggers a maladaptive mechanism including poor ventilatory response, increased sympathetic tone, exaggerated and uneven pulmonary vasoconstriction (pulmonary hypertension), and inadequate production of hormonal mediators (e.g., nitric oxide [NO]) that then lead to capillary leak and pulmonary edema.33 The risk for developing HAPE depends on individual susceptibility, altitude ascent rate, and time spent at the altitude. The incidence of HAPE increases at different heights, ranging from 0.2% to 6% at 4500 m to 2% to 15% at 5500 m.34 Clinical symptoms that precede presentation of pulmonary edema include shortness of breath, nonproductive cough, and difficulty in continuing to ascend to greater heights. such symptoms can easily be mistaken for exhaustion. The symptoms usually appear 2 to 4 days after arriving at a new altitude. It is unusual for HAPE to develop after more than 1 week at the same altitude. When symptoms progress, the patient becomes easily exhausted and may have productive pink sputum. In the later stages, the patient becomes severely hypoxemic, a situation that may be fatal without medical treatment. A favorable outcome depends on early recognition of the patient’s signs and symptoms, using supplementary oxygen, rapid descent to lower altitude, or the use of a hyperbaric chamber. No pharmacologic intervention beyond oxygen has been proven to be beneficial for HAPE, but several pharmacologic agents have been examined. Nifedipine (calcium channel blocker) may aid in both lowering the elevated pulmonary pressure and the systemic resistance. Tadalafil and sildenafil are phosphodiesterase-5 inhibitors acting on the pulmonary vasculature by increasing the amount of available nitric oxide. These agents have been shown to be beneficial for prophylactic treatment of HAPE but have not been examined in the treatment of this condition. Salmeterol, an inhaled β-agonist, has been proposed as a prophylactic drug for HAPE that may also be useful for treatment.
Pregnancy-Related Pulmonary Edema
Pregnancy causes significant hemodynamic changes in the cardiovascular system, including increase in plasma volume, cardiac output, heart rate, and capillary permeability, as well as decreased colloid osmotic pressure. In light of these and other factors, pulmonary edema may occur in pregnant women with preexisting cardiac conditions or abnormalities (cardiomyopathies and valvular disease) or with pregnancy-related abnormalities such as preeclampsia. The incidence of pulmonary edema ranges from 0.08% in normal pregnancies to 3.4% in preeclampsia and up to 5% in preterm labor.35 In a large survey including 62,917 women, the overall incidence of pulmonary edema was 0.08%. Among the pregnant women who developed pulmonary edema, the most common attributable causes or associated conditions were tocolytic use (13 patients [25.5%]), cardiac disease (13 patients [25.5%]), fluid overload (11 patients [21.5%]), and preeclampsia (9 patients [18%]).36 The diagnosis of pulmonary edema was made during the antepartum period in 24 patients (47%), the intrapartum period in 7 (14%), and the postpartum period in 20 (39%). The increased incidence of pulmonary edema in the intra- and postpartum period can be attributed to changes in the plasma colloid pressure. Plasma colloid pressure decreases from about 22 to 16 mm Hg at term after delivery in normal pregnancy, and from 18 to 14 mm Hg postpartum in preeclampsia complicated pregnancies. This reduction is attributed to blood loss and fluid shift due to increased vascular permeability, especially in pregnancies with preeclampsia, and leads to pulmonary edema occurring after delivery.37 Women with preeclampsia are at increased risk for the development of pulmonary edema due to underlying endothelial damage and decreased colloid osmotic pressure, which cause leakage into the pulmonary interstitium or alveolar space.
The development of pulmonary edema associated with pregnancy appears to be influenced by maternal age, parity, and preexisting essential hypertension. In a small study examining the role of echocardiography in the diagnosis of pulmonary edema in the setting of preeclampsia, 25% of the patients had decreased systolic function, and a significant number of the remaining patients had elevated diastolic pressures when compared to other pregnant hypertensive/normotensive women without preeclampsia, thereby indicating that elevated filling pressure may be a part of the pathologic process in preeclampasia.38 Tocolytics are also a major cause for pulmonary edema during pregnancy. Therapy using β-agonists can cause increased hydrostatic pressure and lead to pulmonary edema.39 Pulmonary edema has also been reported after usage of calcium channel blockers as tocolytics.40 As in most patients with pulmonary edema, the mainstay of treatment includes fluid restriction, diuretics, and cessation of tocolytics.
Postobstructive Pulmonary Edema
Postobstructive pulmonary edema (POPE) was first described in 1973 as sudden onset of pulmonary edema following relief of upper airway obstruction. The incidence may be up to 10% of cases after the relief of acute obstruction and up to 40% after relief of chronic obstruction.41 Two types are described: type I POPE follows a sudden, severe episode of upper airway obstruction such as postextubation laryngospasm, epiglottitis, or croup and is seen in strangulation and hanging; type II POPE develops after surgical relief of chronic upper airway obstruction.42 Type I POPE usually develops within 1 hour after the event, but it can be delayed up to 6 hours. In contrast, there is close proximity between the relief of the obstruction and the development of POPE in type II.
The etiology for type I POPE is multifactorial. Negative intrathoracic pressure is caused by inhaling against closed obstruction. This causes increased venous return, decreased cardiac output, and fluid transudation into the alveolar space.43 Risk factors for type I POPE are young age (owing to increased ability to generate increased negative pressure), direct suctioning of the endotracheal tube during thoracotomy, narcotics, short neck, oral or pharyngeal surgery or pathology, vocal cord paralysis, conditions leading to increased capillary-alveolar pressure gradients, endotracheal tube obstruction, and premature extubation. The etiology for type II POPE, which is less frequent than type I, is less clear. It is suggested that the obstructive lesion causes constant positive end-expiratory pressure (PEEP) with increased end-expiratory lung volume. Relief of the obstruction causes immediate reduction of the lung volume that is postulated to result in increased pulmonary permeability and transudation of fluid. The diagnosis usually is suggested by physical findings after surgery of tachypnea, tachycardia, agitation, and frothy pulmonary secretions. The diagnosis is confirmed by x-ray. Most patients will respond quickly to standard therapy with adjunct support of PEEP (5 mm H2O).
Reexpansion Pulmonary Edema
Reexpansion pulmonary edema (REPE) after spontaneous pneumothorax is a rare complication of tube thoracostomy, with reported mortality ranging from 0 to 20%.30,31,44,45 Most patients will present with symptoms as early as 1 hour after thoracostomy, but delayed presentation of up to 24 hours after thoracostomy has also been described. Tachypnea, tachycardia, and hypoxia are the main presenting signs and symptoms. The chest radiograph demonstrates unilateral pulmonary edema, although bilateral pulmonary edema has rarely been reported. In a recent study, many REPE cases were mild and asymptomatic and only diagnosed by computed tomography (CT) of the chest. Most cases will resolve within 24 to 72 hours.
The pathophysiology of REPE is unclear. The main hypothesis is that capillary leak is induced by a postexpansion inflammatory process. During reexpansion, mechanical injury to the alveolar-capillary membrane, together with reperfusion injury from the reinstitution of blood flow, initiates an acute inflammatory process. Predictive factors for REPE are age (20-39 years) and prolonged duration of pneumothorax prior to relief.46 It was also suggested that REPE may be related to the application of negative pressure to the chest tube. No human study has been performed prospectively to determine whether the incidence of REPE would be less if the chest tube is put to water seal only. Unfortunately, REPE can also occur in patients whose lungs are reexpanded without suction. REPE therefore appears to be related to three factors: longer duration of pneumothorax, greater size of the pneumothorax, and a rapid rate of expansion after tube thoracostomy. Controlling for one factor may not prevent the process if one or two of the other factors are present. In lieu of a randomized controlled trial, the American College of Chest Physicians (ACCP) recommends that in the presence of a spontaneous pneumothorax in clinically stable patients with a large (≥30% of the lung field) primary pneumothorax, either a small-bore (14F or smaller) catheter or 16 to 22F chest tube with the tube connected to Heimlich valve or a water-seal device be placed. However, if the lung fails to reexpand, application of negative pressure to the chest tube is deemed appropriate.47
Transfusion-Related Pulmonary Edema
Acute onset of dyspnea shortly after blood transfusion can be attributed to two main etiologies: transfusion-associated cardiac overload (TACO) and immune-mediated ALI resulting from transfusion of plasma-containing products (transfusion-related ALI, or TRALI).48 TRALI was defined by the National Heart, Lung, and Blood Institute Working Group as an ALI that develops within 6 hours after blood transfusion.49 TRALI is considered to be the leading cause for transfusion-related mortality. Virtually all blood products can lead to TRALI, but infusions of whole blood, platelets, packed red blood cells, and fresh frozen plasma are the most commonly identified precipitating causes. Owing to nonuniformity of definitions, the true incidence of TRALI is uncertain, but when uniform definitions are used, the incidence is reported to be 1 case for every 1000 to 2400 units transfused, with equal incidence between men and women and wide age variability.50 Risk factors for TRALI are prolonged storage of blood products, fresh frozen plasma infusion, and underlying conditions such as recent surgery, thrombocytopenia, and massive transfusions. The pathogenesis of TACO is similar to other causes of acute congestive heart failure: volume overload leading to increased central and pulmonary pressures resulting in increased hydrostatic pressure and extravasation of fluid into the alveolar space. The pathogenesis of TRALI is less obvious. Three hypotheses are proposed: (1) antigranulocyte antibodies in the donor’s plasma (or less commonly, in the recipient’s plasma) react with antigens on the recipient’s (or less commonly, donor’s) granulocytes to initiate an inflammatory response within the pulmonary microvasculature; (2) biologically active substances such as lipids and cytokines contained within the transfusions prime granulocytes in the pulmonary vasculature, contributing to increased vascular permeability; or (3) a “two-hit” hypothesis wherein the primary stimulus causes granulocyte sequestration in the pulmonary capillaries, and a secondary stimulus causes the granulocytes to “activate,” damaging the endothelial layer such that fluid and protein leak into the alveolar space. Surgery, infections, and other situations can serve as the initial primer for this process.51
The mainstay of TACO treatment is discontinuation of blood-product transfusion, respiratory support as needed, and diuretics. It has been suggested that subsequent blood products should be infused at a slower rate after the appearance of TACO, but no solid evidence supports this suggestion. TRALI treatment is mainly supportive: mechanical positive-pressure invasive ventilation and high concentrations of oxygen and PEEP. Although some authors have advocated the use of steroids for TRALI, this approach is still considered anecdotal.52 The mortality rate for TRALI varies between 5% and 8%, but rates of up to 47% in critically ill patients have also been reported.48 Most survivors recover completely with appropriate treatment. It is recommended that patients who recover from TRALI should not receive any other blood products from the same donor, but it seems they are not at increased risk for TRALI when receiving blood products from other donors.
Drug Toxicities
Development of pulmonary edema has been linked to a number of drugs and substances.
Opiates
Opiate overdose can induce pulmonary edema due to increased capillary permeability. Interstitial protein content in this setting is similar to the plasma protein content. The pulmonary capillary wedge pressure is generally within the normal range. Direct toxic and hypoxic etiologies have also been suggested as contributing mechanisms in this clinical setting. The incidence of pulmonary edema in patients with heroin overdose is 0.8% to 2.4%,53 with most of the symptoms developing over the first 2 hours of admission. The entire opiate family shares the ability to induce pulmonary edema, and even overdose of codeine has been linked to this condition.54 Most patients require mechanical ventilation to correct severe hypoxia and respond within 24 to 36 hours to supportive care.
Salicylates
Salicylate intoxication can induce pulmonary edema. The mainstay of treatment for aspirin intoxication is volume resuscitation and bicarbonate, which can lead to volume overload and pulmonary edema that cannot be easily differentiated from ALI. Development of pulmonary edema in the setting of aspirin intoxication is an indication for immediate hemodialysis.55
Neurogenic Pulmonary Edema
Acute central nervous system (CNS) injury may lead to a clinical presentation similar to ARDS.56 Symptoms develop within minutes to several hours after the offending injury. Classic signs and symptoms of pulmonary edema include tachycardia, tachypnea, basilar rales on auscultation, and bilateral infiltrates on chest radiograph. Both cardiac output and pulmonary capillary wedge pressure are normal in this situation. These signs and symptoms together with evidence of acute CNS injury establish the diagnosis. The most common causes for neurogenic pulmonary edema (NPE) are epileptic seizures, head injury, and cerebral hemorrhage, but any intracranial or spinal injury can be associated with this condition.56
There are several theories describing the precipitating factors leading to NPE. Excessive stimulation of the autonomic nervous system can result in pulmonary venous vasoconstriction, causing elevations in hydrostatic pressure and extravasation of fluid into the pulmonary interstitium. This mechanism is supported by data showing the ability of α-adrenergic agonists to alleviate pulmonary edema caused by cerebral stimulation in rats.57 Furthermore, rapid elevation of pulmonary venous pressure may cause microvascular injury and excessive capillary permeability, leading to ALI. Two etiologies must be differentiated from NPE in the setting of an intubated head injured patient: aspiration pneumonia and ventilator-associated pneumonia (VAP). The treatment of NPE must first focus on treatment of the offending head/spinal injury. It is essential that hematomas are evacuated, intracranial pressure (ICP) decreased, and convulsions controlled. Other supportive therapies include ventilation that meets the oxygenation needs of the patient, with permissive hypercapnia allowed only in patients with ICP monitoring, and avoidance of high PEEP that may reduce cerebral perfusion. Hemodynamic support should aim to maintain low cardiac filling pressures without compromising cerebral perfusion. Invasive hemodynamic monitoring may be required. The exact place of α- and β-adrenergic agents in the therapy of NPE is not established. Most NPE episodes will resolve within 48 to 72 hours.
Other Noncardiogenic Etiologies of Acute Pulmonary Edema
Massive pulmonary embolism (PE) can cause pulmonary edema due to elevated hydrostatic pressure and injury to adjacent pleural and pulmonary vasculature. Viral infections can also induce pulmonary edema, as demonstrated in severe cases of Hanta virus infection.58 Other viruses, including enteroviruses and coronavirus, can also lead to pulmonary edema.
There are reports of pulmonary edema in trained athletes after strenuous exercise such as marathon running or swimming. The theories that explain this phenomenon point to preexisting ventilation/perfusion mismatch in combination with increased cardiac output, causing elevated hydrostatic pressure that leads to pulmonary edema.59,60 Cold water immersion can induce pulmonary edema by both increasing cardiac output and elevating pulmonary vascular resistance and pressures.



Full access? Get Clinical Tree


