Prostanoids and Other Inflammatory Mediators
Christian Waeber
Introduced by Bayer in 1899, acetylsalicylic acid (aspirin [ASA]) is today the most extensively prescribed analgesic, antipyretic, and anti-inflammatory agent, with 120 billion tablets consumed annually (68). The efficacy of ASA (and other nonsteroidal anti-inflammatory drugs [NSAIDs]) for headache relief has been extensively assessed in placebo-controlled studies (33,40,49,62,73). Sir John Vane was awarded the Nobel Prize for Physiology or Medicine in 1982 for his discovery that aspirin’s major therapeutic and adverse effects could be explained by inhibition of prostaglandin synthesis by inactivation of the key enzyme, cyclooxygenase (COX) (67). However, despite being among the most commonly used antimigraine drugs, the precise mechanism of action of these agents in migraine headache therapy is still not clear.
We now know that at least three COX isoforms exist (Figure 18-1). COX-1 is the constitutive enzyme producing prostaglandin and thromboxanes involved in physiologic activities like cytoprotection and platelet aggregation, whereas COX-2 appears to be preferentially expressed in inflamed tissues. Recently, a COX-1 splice variant, named COX-3, has been cloned and found to be abundantly expressed in mature brain and spinal cord (10). COX-3 inhibition in the brain may be responsible for analgesic and antipyretic effects of certain NSAIDs. Whereas ASA inhibits all three COX isoforms, a new class of inhibitors termed coxibs selectively inhibit COX-2. Acetaminophen blocks mostly COX-3. ASA is unique because it inactivates COX by irreversible acetylation of serine residue in the active site of the enzyme; other NSAIDs are competitive reversible inhibitors of COX. Prostaglandins sensitize free nerve endings (pain receptors) to numerous inflammatory mediators and, when injected directly into the brain, induce fever and pain. ASA, by inhibiting all isoforms of COX, can be considered both a peripherally and centrally acting analgesic. The central mechanism of action is supported by autoradiographic studies that showed high-affinity binding of ASA to nociceptive structures in the brain (26). Additionally, in animal studies, intravenous ASA inhibits central trigeminal neurons in the dorsal horn of the upper cervical spinal cord after stimulation of the sagittal superior sinus (35).
Because of the established efficacy of NSAIDs in migraine therapy, prostanoids and their potential targets are highly relevant to migraine pathophysiology. However, various other mediators are important for inflammatory processes, and might provide additional therapeutic opportunities in migraine. They are therefore discussed in the following section.
INFLAMMATORY MEDIATORS
Commonly, inflammation occurs as a defensive response to invasion of the host by foreign, particularly microbial, material. Responses to mechanical trauma, toxins, and neoplasia may also result in inflammatory reactions. Microscopically, inflammation involves a complex series of events, including dilation of arterioles, capillaries, and venules, with increased permeability and blood flow, as well as exudation of fluids, including plasma proteins. The accumulation and subsequent activation of leukocytes are central events in the pathogenesis of most forms of inflammation. The development of inflammatory reactions is controlled by various mediators, including free radicals, the complement system, kinins, cytokines, and prostanoids.
Free radicals are atoms or molecules that readily react with other cellular structures because they contain unpaired electrons. Reactive oxygen species (ROS) such as hydrogen peroxide, superoxide, and hydroxyl species, are normal byproducts of cellular electron transfer reaction, ordinary metabolic processes, and immune system responses (the secretion of reactive oxygen and
nitrogen free radical species by inflammatory cells is a major mechanism for attacking foreign substances). Freeradical-generating substances can also be found in food or drugs. ROS are important in inflammation and may play a role in nociceptor activation. For instance, hydrogen peroxide has been shown to enhance the effects of other inflammatory mediators (bradykinin, prostaglandins), and nitric oxide ([NO] a reactive nitrogen species) induces a delayed burning pain upon intradermal injection (29). NO donors can activate sensory fibers directly, causing the release of CGRP (72). There is also strong evidence that NO is involved in the migraine pathogenesis (see below).
nitrogen free radical species by inflammatory cells is a major mechanism for attacking foreign substances). Freeradical-generating substances can also be found in food or drugs. ROS are important in inflammation and may play a role in nociceptor activation. For instance, hydrogen peroxide has been shown to enhance the effects of other inflammatory mediators (bradykinin, prostaglandins), and nitric oxide ([NO] a reactive nitrogen species) induces a delayed burning pain upon intradermal injection (29). NO donors can activate sensory fibers directly, causing the release of CGRP (72). There is also strong evidence that NO is involved in the migraine pathogenesis (see below).
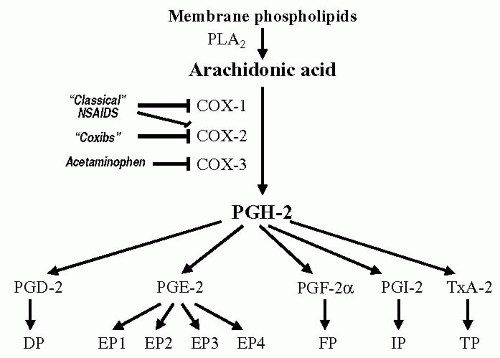 FIGURE 18-1. Summary of biosynthetic routes to the major prostaglandins and thromboxane A2. Arachidonic acid is generated from membrane phospholipids via the action of two types of phospholipase A2 (a secreted 14 kilodalton form, and the type IV cytosolic 85 kilodalton isoform [6]). Arachidonic acid is the converted to the prostaglandin endoperoxides PGG2 (not shown) and PGH2 by two enzymes, COX-1 and COX-2. A COX-1 splice variant, named COX-3, has been cloned and found to be abundantly expressed in mature brain and spinal cord (10). PGH2 spontaneously decomposes in aqueous solution to form a mixture of PGD2, PGE2, and PGF2. However, distinct enzymes catalyze the formation of each of these prostaglandins, as well as TXA2 and PGI2. The cellular expression pattern of each of these enzymes may influence the type of prostaglandin produced by a particular cell. Prostaglandins are liberated from cells and bind to a family of G protein coupled receptors (see Table 18-2). |
The complement comprises several families of proteins activated in sequence when cells are exposed to a foreign substance. Once the proteins are activated, nine of these proteins come together to form the membrane attack complex (MAC). When assembled on a cell membrane, MAC forms a ring-like structure that allows the movement of ions and small molecules into and out of the cell, resulting in cell damage. In addition to these toxic effects, complement anaphylatoxins C3a and C5a initiate local inflammatory responses and various complement proteins also activate phagocytic and endothelial cells. Interestingly, plasma completely loses its ability to excite trigeminal neurons after heat inactivation, suggesting that the complement system may be involved in the excitatory nociceptive transmission in the trigeminal system (21).
Kinins (bradykinin, kallidin) exert a number of proinflammatory effects via two distinct receptors (B1 and B2), including the release of prostanoids, cytokines, and free radicals from a variety of cells. They also stimulate postganglionic sympathetic neurons, degranulate mast cells to release various inflammatory mediators, and cause plasma extravasation by contraction of vascular endothelial cells. They are potent algogenic substances and induce pain by directly stimulating nociceptors and sensitizing them to mechanical stimuli (69).
Cytokines are signaling proteins that are secreted by various types of immune cells. The central role of cytokines is to control the direction, amplitude, and duration of the inflammatory response. Pro-inflammatory cytokines (interleukin [IL]-1, IL-6, tumor necrosis factor [TNF]-α, transforming growth factor-β) are produced predominantly by activated immune cells and are involved in the amplification of inflammatory reactions. Anti-inflammatory cytokines (IL-4, IL-10, and IL -13) are involved in the reduction of inflammatory reactions. IL-1β, IL-6, IL-8, and TNF-α can indirectly induce powerful hyperalgesia by causing prostanoid release, increasing the expression of nerve growth factor (NGF) and bradykinin receptors, or affecting sympathetic fibers (15,16,34).
Prostanoids are a group of lipid mediators that consist of the prostaglandins and thromboxanes. In response to cell stimulation, prostanoids are synthesized by the COX pathway from arachidonic acid released from membrane phospholipids by the actions of phospholipases. Prostanoids, once formed, are quickly released to the outside of cells. Because of their chemical and metabolic instability, prostanoids are believed to act in the vicinity of their sites of production. Thus, they are “short-range hormones,” maintaining local homeostasis in a variety of tissues and cells. Although some prostaglandins have anti-inflammatory effects (they decrease inflammation, increase oxygen flow, prevent cell aggregation, and decrease pain), others are known to have pro-inflammatory effects.
MIGRAINE AS AN INFLAMMATORY DISORDER
Despite considerable research into the pathogenesis of idiopathic headaches such as migraine, their underlying pathophysiologic mechanisms remain poorly understood. A recent meta-analysis of clinical literature published between 1966 and 1999 found only about 45 clinical investigations reporting alterations of immune function in migraine patients (37). Changes of serum levels of complement and immunoglobulins, histamine, cytokines, and
immune cells were reported in some of these studies, but in most cases were not corroborated by others. The inflammatory processes possibly involved in migraine are therefore unlikely to be of immune origin.
immune cells were reported in some of these studies, but in most cases were not corroborated by others. The inflammatory processes possibly involved in migraine are therefore unlikely to be of immune origin.
According to modern theories of migraine pathogenesis, initial activation of intracranial perivascular sensory fibers supplying the dura mater might result from exposure to endogenous (brain-generated or bloodborne) algogenic chemicals. Activation of meningeal nociceptors causes these sensory fibers to release neuropeptides, including substance P and calcitonin gene-related peptide (CGRP) (43). This initiates neurogenic inflammation in the dura mater, leading to a further secretion of inflammatory agents such as serotonin, histamine, bradykinin, and prostaglandins. After application of a cocktail of these inflammatory mediators to the dural sinuses, rat meningeal primary afferent neurons rapidly become mechanically hypersensitive (i.e., neurons that showed no or only minimal response to a small dural indentation before the chemical irritation show a strong response minutes after application of these inflammatory mediators) (63). Similarly, this cocktail can cause central trigeminal neurons receiving convergent input from the dura and the skin to lower their thresholds to mechanical stimulation of the dura and to mechanical and thermal stimulation of the skin. Application of lidocaine to the dura abolishes the response to dural stimulation, but has minimal effect on the increased response to cutaneous stimulation, suggesting involvement of a central mechanism in maintaining the sensitized state, which can last up to 10 hours (8). On the basis of these studies, Burstein (7) has proposed that (a) sensitization of both peripheral and central trigeminovascular neurons accounts for the intracranial hypersensitivity observed in migraineurs, and (b) sensitization of central but not peripheral trigeminal neurons is responsible for the extracranial hypersensitivity (extracranial tenderness and cutaneous allodynia) often seen in these patients.
Nitric oxide, a short-lived vasodilator and reactive nitrogen species, has been implicated in the genesis of migraine. Indeed, headache is a well-known side effect of NO donors such as nitroglycerin or sodium nitroprusside. Intravenous infusion of nitroglycerin causes migraineurs, and not control subjects, to develop a delayed migraine attack (47). Furthermore, the NO synthase inhibitor NG-methylarginine (L-NMMA) effectively improves spontaneous migraine headaches and associated symptoms such as phono- and photophobia (39). These human studies implicate NO in the genesis of headache. In addition, there is also abundant evidence about NO in pain generation and maintenance derived from animal models (41).
Based on these data indicating that NO may participate in the early and late phases of a migraine attack, several groups have studied the effects of NO (administered as the NO donor nitroglycerin) on cephalic structures (38,53,64). Recently, nitroglycerin infusion has been shown to cause a delayed expression of inducible nitric oxide synthase (iNOS) in rat meningeal macrophages (56,57). This iNOS induction is preceded by the appearance of IL-1β in the dura mater, and is followed by mast cell degranulation, IL-6 expression, and plasma protein extravasation (blocked by administration of an iNOS inhibitor), indicating the occurrence of nitroglycerin-induced inflammatory events in the meninges. It is likely that a similar inflammatory response occurs in more spontaneous types of migraine attacks.
RECEPTORS FOR INFLAMMATORY MEDIATORS
G protein-coupled receptors (GPCRs) are the most important target for the pharmaceutical industry, as is indicated by the fact that they are the site of action of 52% of all medicines available today (48). This is probably because of the relative ease of designing low-molecular-weight agents blocking the interaction of small transmitter molecules and their receptors. GPCRs for inflammatory mediators (prostaglandin and kinins, but also chemokines) are important mediators of peripheral sensitization. Because of their potential importance to migraine drug therapy, their pharmacology is reviewed in this section.
Kinin Receptors
Kinins are 9-11 amino acid peptides acting on blood vessels and involved in cardiovascular regulation, inflammation, and pain. Their effects are mediated by two GPCR subtypes: B2 (constitutively expressed) and B1 receptors (inducible and upregulated in the presence of cytokines, endotoxins or tissue injury; Table 18-1). These receptors have been defined based on pharmacologic criteria: bradykinin, kallidin, and T-kinin are endogenous agonists for B2 receptors, and Des-Arg9-bradykinin and Des-Arg10-kallidin prefer B1 receptors (54,55). B1 receptors exert both protective (e.g., in multiple sclerosis and septic shock) and negative effects (pain, edema, and inflammation) and play a major role in the chronic phase of the pain and inflammatory responses. In contrast, B2 receptors are important mediators of the acute phase of inflammation (arterial relaxation, venoconstriction, increased permeability) and of somatic and visceral pain. Autoradiographic studies show that B2 receptor binding sites predominate in the superficial laminae of the dorsal horn, particularly on the terminals of Aδ and C fibers (14), and intrathecal bradykinin administration results in both antinociception (via activation of B2 receptors on the terminals of sensory fibers) and nociception (through the release of noradrenaline from inhibitory neurons projecting to the dorsal horn) (14). It is therefore
likely that the stimulation of central B2 receptors plays an important role in migraine pain.
likely that the stimulation of central B2 receptors plays an important role in migraine pain.
Related posts:
 Principles of Clinical Pharmacology, Randomized Controlled Clinical Trials, and Evidence-Based Medicine in Headache
Principles of Clinical Pharmacology, Randomized Controlled Clinical Trials, and Evidence-Based Medicine in Headache
 Nitric Oxide
Nitric Oxide
 The Migraines: Introduction
The Migraines: Introduction
 Channelopathies and Their Possible Relation to Migraines
Channelopathies and Their Possible Relation to Migraines
 Autonomic Dysfunction in Migraines
Autonomic Dysfunction in Migraines
 Psychological and Behavioral Treatments of Migraines
Psychological and Behavioral Treatments of Migraines

Full access? Get Clinical Tree


