Principles of Clinical Pharmacology, Randomized Controlled Clinical Trials, and Evidence-Based Medicine in Headache
Carl G. H. Dahlöf
Paul Rolan
Peer Tfelt-Hansen
The clinical pharmacology of a drug includes both pharmacokinetics—what the body does to the drug—and pharmacodynamics—what the drug does to the body. In this chapter, a brief account of these aspects of clinical pharmacology relevant to migraine and its treatment will be given, which should help the reader to understand the expressions used in later chapters addressing headache therapy. This chapter also includes a section on the evaluation of the quality of the methods and results in randomized controlled clinical trials in migraine in the perspective of the use of evidence-based medicine in clinical practice.
PHARMACOKINETICS
Pharmacokinetics is the aspect of clinical pharmacology dealing with the degree of drug absorption, the time taken to achieve a peak plasma concentration, the degree of binding to plasma proteins, its distribution from plasma to tissue site of action, the extent of its metabolism, and the pattern of excretion. The absorption, distribution, biotransformation, and excretion of a drug all involve its passage across cell membranes (2). Important characteristics of a drug are its molecular size and shape, solubility at the site of its absorption, degree of ionization, and relative lipid solubility of its ionized and nonionized forms.
Absorption
The degree of absorption of a drug depends on its route of administration: oral, sublingual, subcutaneous, rectal, or inhalational. Absorption from the gastrointestinal tract is governed by factors that are generally applicable, such as surface area for absorption, blood flow to the site of absorption, physical state of the drug, and its concentration at the site of absorption. Any factor that accelerates gastric emptying likely increases the rate of drug absorption, whereas any factor that delays gastric emptying probably has the opposite effect, regardless of the characteristics of the drug. During migraine attacks, gastrointestinal stasis may impair the absorption of aspirin and tolfenamic acid, and the absorption can be normalized by administering the antiemetic metoclopramide, which promotes gastrointestinal motility (4,32,33), but the neuroleptictype antiemetic thiethylperazine does not have this effect (33).
The bioavailability of a drug is an estimate of the amount of intact drug entering the systemic circulation after administration by the intended route and therefore is determined by absorption, and first-pass metabolism. The bioavailability after a certain route of administration is calculated by comparing the area under the drug plasma concentration curve (AUC) with that of the AUC after intravenous administration, expressed as a percentage. That bioavailability is not the same as the amount of a drug absorbed can be exemplified by ergotamine. Sixty-six percent of ergotamine is absorbed after oral administration, but the bioavailability is less than 1%, as a result of extensive first-pass metabolism in the liver (see Chapter 51). If the concentration of a drug in plasma is plotted against time after administration, a curve is outlined, the peak height (Cmax) and the time taken to reach the peak (tmax). Their values depend on absorption, but less obviously, elimination.
Drug Distribution
Patterns of drug distribution reflect certain physiologic factors and physicochemical properties of drugs. An initial phase of distribution may be distinguished that reflects cardiac output and regional blood flow. The most important facet of a drug’s distribution is its movement into bodily tissues, especially whether or not the drug is able to cross the blood-brain barrier. Cerebral blood flow is the only limitation to penetration into the central nervous system (CNS) by highly lipid-soluble drugs. The rate of diffusion of drugs with increasing polarity into the CNS is proportional to the lipid solubility of the nonionized molecules.
The apparent space in the body available to contain the drug is called the volume of distribution. Dividing the amount of drug in the body by the plasma concentration yields a volume of distribution. A large volume of distribution indicates that little of the drug is left in the central compartment and that most of the drug has been distributed to other peripheral compartments.
METABOLISM AND EXCRETION
Generally, the aim of biotransformation is to turn lipophilic drugs into more hydrophilic metabolites that are readily excreted from the body. Many drugs are in this way inactivated by liver metabolism, although formation of active metabolites can take place as an intermediate step of biotransformation.
The kidney is the most important organ for elimination of drugs and their metabolites. The elimination half-life (t1/2) of a drug from plasma (time taken for plasma concentration of a drug to halve) is important as a guide for estimating times for elimination of drugs from the body and, during multiple dosing, as a guide for calculating the rate of accumulation of the drug. A large volume of distribution means that little of the drug is available for metabolism/excretion in the central compartment, and this generally explains the longer elimination half-life of the drug.
Plasma Concentration Measurements
If we could measure the concentrations of a drug at its site of action, the more likely we would be able to link those concentrations to a clinical response; but this is not possible in migraine. The next best alternative is to try to relate plasma drug concentrations with clinical or pharmacologic effects; but so far this approach has not been successful in migraine either, even when the concentrations of active metabolites also have been taken into account (3). The only current use for measuring drug levels is to check for compliance and with lithium no safeguard against overdosing.
PHARMACODYNAMICS
Pharmacodynamics is the study of pharmacologic effects and mechanisms of action once a drug has reached its site(s) of action from the blood stream after absorption.
Mode of Drug Action
Most drugs act specifically as agonists or antagonists of receptors, as uptake inhibitors, ion channel modifiers, or enzyme inhibitors. The particular mechanisms relevant to drug action in headache therapy are addressed in those sections of this book dealing with individual drugs. Suffice it to say that nearly all of the drugs used in the treatment or prevention of migraine act on specific receptors, especially those that are the natural receptors for neurotransmitters released by serotonergic, adrenergic, and dopaminergic neurons or receptors for excitatory amino acids and neuropeptides. Most drugs act specifically as agonists or antagonists of receptors, as uptake inhibitors, ion channel modifiers, or enzyme inhibitors. The particular mechanisms relevant to drug action in headache therapy are addressed in those sections of this book dealing with individual drugs. Suffice it to say that nearly all of the drugs used in the treatment or prevention of migraine act on specific receptors, especially those that are the natural receptors for neurotransmitters released by serotonergic, adrenergic, and dopaminergic neurons or receptors for excitatory amino acids and neuropeptides. The serotonergic and other adrenoceptors are located not only on structures in the head, brain, vessels, and other tissues, but in peripheral structures, including blood vessels throughout the body, the heart, and organs containing smooth muscle, such as the gastrointestinal tract, tracheobronchial tree, and bladder. It follows, therefore, that a drug action directed at a particular specific receptor in the head also affects those peripheral structures possessing the same receptor species. Neurotransmitter pharmacology is complicated even more by the fact that several specific receptor sub-types exist for the same endogenous transmitter substance (at least 14, for instance, have been claimed in the case of 5-hydroxytryptamine [5-HT]) (9,10,23,30). Furthermore, receptors for entirely different substances sometimes possess significant structural homology, which explains why certain drugs affect (stimulate or block) more than one type of receptor. Ergotamine, for example, is a powerful agonist on 5-HT1A, 5-HT1B, 5-HT1C, 5-HT1D, 5-HT2A, and 5-HT2C receptors and dopamine receptors. In addition to that, ergotamine also has a mixed agonist/antagonist pharmacology at the α1- and α2-adrenoceptors (25).
Dose-Response Relationship
The animal pharmacologist may be able to observe changes in the magnitude of a drug response over a wide range of drug concentrations to determine the complete
dose-response relationship. If the response, expressed as a percentage of the maximal response that an agonist drug is capable of inducing, is plotted against the logarithm of the dose (concentration), a sigmoid curve usually results, with points between 20% and 80% falling on a straight line. However, not all agonist drugs that act on the same receptors can elicit the same maximal response. Such differences have been explained as the drugs possessing different intrinsic activities (or efficacies). Thus, a drug (X) with high intrinsic activity capable of causing the maximal possible tissue response is a full agonist, whereas one (Y) with low intrinsic activity that can induce a less than maximal response, even with the highest doses possible when acting on the same receptor as drug X, is a partial agonist (Fig. 7-1A). Because it occupies receptors that would otherwise be free for activation by a full agonist, a partial agonist behaves as a partial antagonist in the presence of a full agonist.
dose-response relationship. If the response, expressed as a percentage of the maximal response that an agonist drug is capable of inducing, is plotted against the logarithm of the dose (concentration), a sigmoid curve usually results, with points between 20% and 80% falling on a straight line. However, not all agonist drugs that act on the same receptors can elicit the same maximal response. Such differences have been explained as the drugs possessing different intrinsic activities (or efficacies). Thus, a drug (X) with high intrinsic activity capable of causing the maximal possible tissue response is a full agonist, whereas one (Y) with low intrinsic activity that can induce a less than maximal response, even with the highest doses possible when acting on the same receptor as drug X, is a partial agonist (Fig. 7-1A). Because it occupies receptors that would otherwise be free for activation by a full agonist, a partial agonist behaves as a partial antagonist in the presence of a full agonist.
When antagonists occupy receptors, they induce no response and therefore have no intrinsic activity. They merely prevent agonists from inducing their responses. In the presence of a competitive antagonist, the log dose-response curve to the agonist is displaced in a parallel manner to the right of the curve in its absence (Fig. 7-1B). An antagonist that acts noncompetitively binds irreversibly to receptors, and the maximal response of the tissue to the corresponding agonist can no longer be achieved (Fig. 7-1C) unless the tissue possesses excess or spare receptors. In this case a full agonist can cause a maximal response by stimulating only a small proportion of the total number of receptors and induces a maximal tissue response even in the presence of a noncompetitive antagonist until the latter leaves too few receptors to enable the agonist to cause a maximal response.
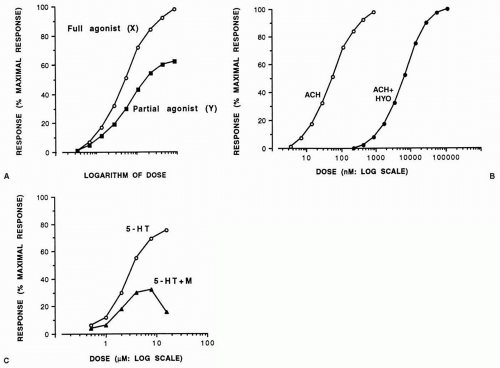 FIGURE 7-1. (A) Log dose-response curves to full (X) and partial (Y) agonists. The ordinate is the percentage maximal response. (B) Competitive antagonism. The example shown is of a full log dose-contractile response curve to acetylcholine on the guinea pig isolated ileum in the absence (ACH) and presence (ACH + HYO) of the muscarinic cholinoceptor antagonist hyoscine (0.33 μmol). The ordinate is the percentage maximal response to acetylcholine. (C) Noncompetitive antagonism. The example shown is of a full log dose-contractile response curve to 5-HT on the guinea pig isolated ileum in the absence (5-HT) and presence (5-HT + M) of the nicotinic cholinoceptor antagonist mecanylamine (25μmol). The ordinate is the percentage maximal response to acetylcholine, from which it can be seen that maximal 5-HT receptor activation leads to a smaller response of the ileum than maximal acetylcholine receptor activation. |
Related posts:







Full access? Get Clinical Tree


