Chapter 5 Physiology of the Airway
I Normal Respiratory Physiology (Nonanesthetized)
A Gravity-Determined Distribution of Perfusion and Ventilation
1 Distribution of Pulmonary Perfusion
Contraction of the right ventricle imparts kinetic energy to the blood in the main pulmonary artery. As this energy is dissipated in climbing a vertical hydrostatic gradient, the absolute pressure in the pulmonary artery (Ppa) decreases by 1 cm H2O per centimeter of vertical distance up the lung (Fig. 5-1). At some height above the heart, Ppa becomes zero (i.e., equal to atmospheric pressure), and still higher in the lung, Ppa becomes negative.1 In this region, then, alveolar pressure (PA) exceeds Ppa and pulmonary venous pressure (Ppv), which is very negative at this vertical height. Because the pressure outside the vessels is greater than the pressure inside the vessels, the vessels in this region of the lung are collapsed, and no blood flow occurs; this is known as zone 1 (PA > Ppa > Ppv). Because there is no blood flow, no gas exchange is possible, and the region functions as alveolar dead space, or wasted ventilation. Little or no zone 1 exists in the lung under normal conditions,2 but the amount of zone 1 lung may be greatly increased if Ppa is reduced, as in oligemic shock, or if PA is increased, as in the application of excessively large tidal volumes (VT) or levels of positive end-expiratory pressure (PEEP) during positive-pressure ventilation.
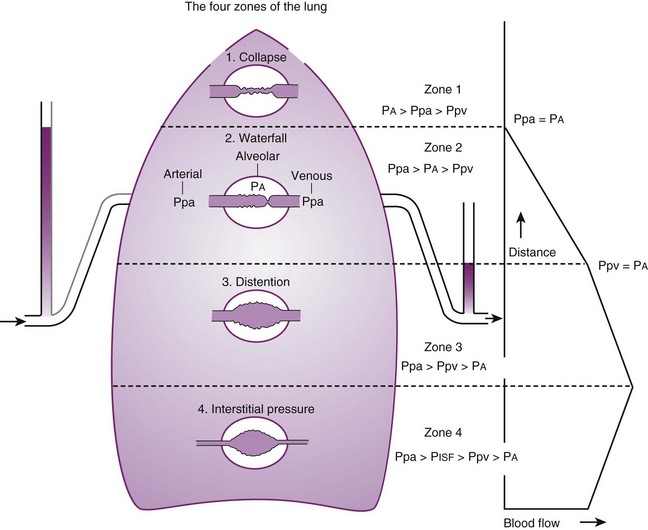
Further down the lung, absolute Ppa becomes positive, and blood flow begins when Ppa exceeds PA (zone 2, Ppa > PA > Ppv). At this vertical level in the lung, PA exceeds Ppv, and blood flow is determined by the mean Ppa − PA difference rather than by the more conventional Ppa − Ppv difference (see later discussion).3 In zone 2, the relationship between blood flow and alveolar pressure has the same physical characteristics as a waterfall flowing over a dam. The height of the upstream river (before reaching the dam) is equivalent to Ppa, and the height of the dam is equivalent to PA. The rate of water flow over the dam is proportional to only the difference between the height of the upstream river and the dam (Ppa − PA), and it does not matter how far below the dam the downstream riverbed (Ppv) is. This phenomenon has various names, including the waterfall, Starling resistor, weir (dam made by beavers), and sluice effect. Because mean Ppa increases down this region of the lung but mean PA is relatively constant, the mean driving pressure (Ppa − PA) increases linearly, and therefore mean blood flow increases linearly as one descends down this portion of the lung. However, respiration and pulmonary blood flow are cyclic phenomena. Therefore, absolute instantaneous Ppa, Ppv, and PA are changing continuously, and the relationships among Ppa, Ppv, and PA are dynamically determined by the phase lags between the cardiac and respiratory cycles. Consequently, a given point in zone 2 may actually be in either a zone 1 or a zone 3 condition at a given moment, depending on whether the patient is in respiratory systole or diastole or in cardiac systole or diastole.
Finally, whenever pulmonary vascular pressures (Ppa) are extremely high, as they are in a severely volume-overloaded patient, in a severely restricted and constricted pulmonary vascular bed, in an extremely dependent lung (far below the vertical level of the left atrium), and in patients with pulmonary embolism or mitral stenosis, fluid can transude out of the pulmonary vessels into the pulmonary interstitial compartment. In addition, pulmonary interstitial edema can be caused by extremely negative Ppl and perivascular hydrostatic pressure, such as may occur in a vigorously spontaneously breathing patient with an obstructed airway due to laryngospasm (most commonly) or upper airway masses (e.g., tumors, hematoma, abscess, edema), strangulation, infectious processes (e.g., epiglottitis, pharyngitis, croup), or vocal cord paralysis; with rapid reexpansion of lung; or with the application of very negative Ppl during thoracentesis.4,5 Transuded pulmonary interstitial fluid can significantly alter the distribution of pulmonary blood flow.
When the flow of fluid into the interstitial space is excessive and the fluid cannot be cleared adequately by the lymphatics, it accumulates in the interstitial connective tissue compartment around the large vessels and airways and forms peribronchial and periarteriolar edema fluid cuffs. The transuded pulmonary interstitial fluid fills the pulmonary interstitial space and may eliminate the normally present negative and radially expanding interstitial tension on the extra-alveolar pulmonary vessels. Expansion of the pulmonary interstitial space by fluid causes pulmonary interstitial pressure (PISF) to become positive and to exceed Ppv (zone 4, Ppa > PISF > Ppv > PA).6,7 In addition, the vascular resistance of extra-alveolar vessels may be increased at a very low lung volume (i.e., residual volume); at such volumes, the tethering action of the pulmonary tissue on the vessels is also lost, and as a result, PISF increases positively (see later discussion of lung volume).8,9 Consequently, zone 4 blood flow is governed by the arteriointerstitial pressure difference (Ppa − PISF), which is less than the Ppa − Ppv difference, and therefore zone 4 blood flow is less than zone 3 blood flow. In summary, zone 4 is a region of the lung from which a large amount of fluid has transuded into the pulmonary interstitial compartment or is possibly at a very low lung volume. Both these circumstances produce positive interstitial pressure, which causes compression of extra-alveolar vessels, increased extra-alveolar vascular resistance, and decreased regional blood flow.
It should be evident that as Ppa and Ppv increase, three important changes take place in the pulmonary circulation—namely, recruitment or opening of previously unperfused vessels, distention or widening of previously perfused vessels, and transudation of fluid from very distended vessels.10,11 Thus, as mean Ppa increases, zone 1 arteries may become zone 2 arteries, and as mean Ppv increases, zone 2 veins may become zone 3 veins. The increase in both mean Ppa and Ppv distends zone 3 vessels according to their compliance and decreases the resistance to flow through them. Zone 3 vessels may become so distended that they leak fluid and become converted to zone 4 vessels. In general, pulmonary capillary recruitment is the principal change as Ppa and Ppv increase from low to moderate levels, distention is the principal change as Ppa and Ppv increase from moderate to high levels, and transudation is the principal change when Ppa and Ppv increase from high to very high levels.
2 Distribution of Ventilation
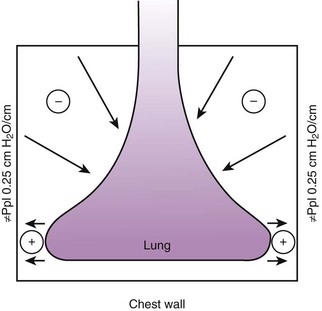
Gravity also causes differences in vertical Ppl, which in turn causes differences in regional alveolar volume, compliance, and ventilation. The vertical gradient of Ppl can best be understood by imagining the lung as a plastic bag filled with semifluid contents; in other words, it is a viscoelastic structure. Without the presence of a supporting chest wall, the effect of gravity on the contents of the bag would cause the bag to bulge outward at the bottom and inward at the top (i.e., it would assume a globular shape). Inside the supporting chest wall, the lung cannot assume a globular shape. However, gravity still exerts a force on the lung to assume a globular shape; this force creates relatively more negative pressure at the top of the pleural space (where the lung pulls away from the chest wall) and relatively more positive pressure at the bottom of the lung (where the lung is compressed against the chest wall) (Fig. 5-2). The density of the lung determines the magnitude of this pressure gradient. Because the lung has about one fourth the density of water, the gradient of Ppl (in cm H2O) is about one fourth the height of the upright lung (30 cm). Thus, Ppl increases positively by 30/4 = 7.5 cm H2O from the top to the bottom of the lung.12
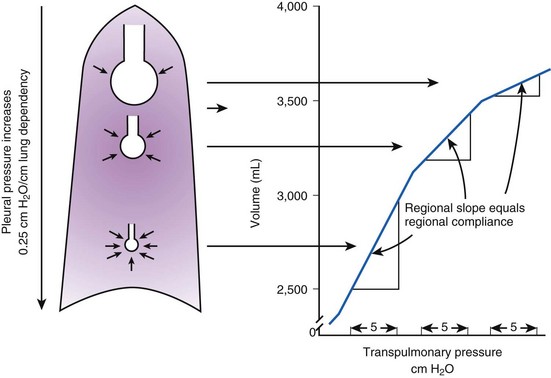
Because PA is the same throughout the lung, the Ppl gradient causes regional differences in transpulmonary distending pressure (PA − Ppl). Ppl is most positive (least negative) in the dependent basilar lung regions, so alveoli in these regions are more compressed and are therefore considerably smaller than the superior, relatively noncompressed apical alveoli. (The volume difference is approximately fourfold.)13 If regional differences in alveolar volume are translated to a pressure-volume (compliance) curve for normal lung (Fig. 5-3), the dependent small alveoli are on the midportion, and the nondependent large alveoli are on the upper portion of the S-shaped compliance curve. Because the different regional slopes of the composite curve are equal to the different regional lung compliance values, dependent alveoli are relatively compliant (steep slope), and nondependent alveoli are relatively noncompliant (flat slope). Therefore, most of the VT is preferentially distributed to dependent alveoli which expand more per unit pressure change than the nondependent alveoli.
3 The Ventilation-Perfusion Ratio
Blood flow and ventilation (both shown on the left vertical axis of Fig. 5-4) increase linearly with distance down the normal upright lung (horizontal axis, reverse polarity).14 Because blood flow increases from a very low value and more rapidly than ventilation does with distance down the lung, the ventilation-perfusion ratio (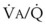 , right vertical axis of Fig. 5-4) decreases rapidly at first and then more slowly.
, right vertical axis of Fig. 5-4) decreases rapidly at first and then more slowly.
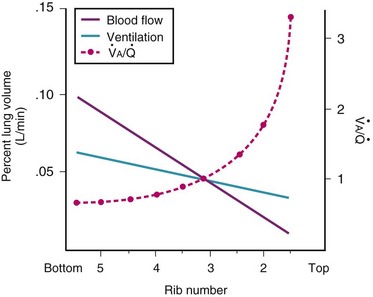
Figure 5-4 Distribution of ventilation and blood flow (left vertical axis) and the ventilation-perfusion ratio (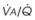 , right vertical axis) in normal upright lung. Both blood flow and ventilation are expressed in liters per minute per percentage of alveolar volume and have been drawn as smoothed-out linear functions of vertical height. The closed circles mark the
, right vertical axis) in normal upright lung. Both blood flow and ventilation are expressed in liters per minute per percentage of alveolar volume and have been drawn as smoothed-out linear functions of vertical height. The closed circles mark the  ratios of horizontal lung slices (three of which are shown in Fig. 5-5). A cardiac output of 6 L/min and a total minute ventilation of 5.1 L/min were assumed.
ratios of horizontal lung slices (three of which are shown in Fig. 5-5). A cardiac output of 6 L/min and a total minute ventilation of 5.1 L/min were assumed.
(Redrawn with modification from West JB: Ventilation/Blood flow and gas exchange, ed 4, Oxford, 1970, Blackwell Scientific.)
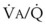 best expresses the amount of ventilation relative to perfusion in any given lung region. For example, alveoli at the base of the lung are overperfused in relation to their ventilation (
best expresses the amount of ventilation relative to perfusion in any given lung region. For example, alveoli at the base of the lung are overperfused in relation to their ventilation (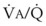 < 1). Figure 5-5 shows the calculated ventilation (
< 1). Figure 5-5 shows the calculated ventilation (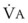 ) and blood flow (
) and blood flow (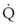 ), the
), the 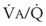 ratio, and the alveolar partial pressures of oxygen (PAO2) and carbon dioxide (PACO2) for horizontal slices from the top (7% of lung volume), middle (11% of lung volume), and bottom (13% of lung volume) of the lung.15 PAO2 increases by more than 40 mm Hg, from 89 mm Hg at the base to 132 mm Hg at the apex, whereas PCO2 decreases by 14 mm Hg, from 42 mm Hg at the bottom to 28 mm Hg at the top. Therefore, in keeping with the regional
ratio, and the alveolar partial pressures of oxygen (PAO2) and carbon dioxide (PACO2) for horizontal slices from the top (7% of lung volume), middle (11% of lung volume), and bottom (13% of lung volume) of the lung.15 PAO2 increases by more than 40 mm Hg, from 89 mm Hg at the base to 132 mm Hg at the apex, whereas PCO2 decreases by 14 mm Hg, from 42 mm Hg at the bottom to 28 mm Hg at the top. Therefore, in keeping with the regional 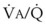 ratio, the bottom of the lung is relatively hypoxic and hypercapnic compared with the top of the lung.
ratio, the bottom of the lung is relatively hypoxic and hypercapnic compared with the top of the lung.
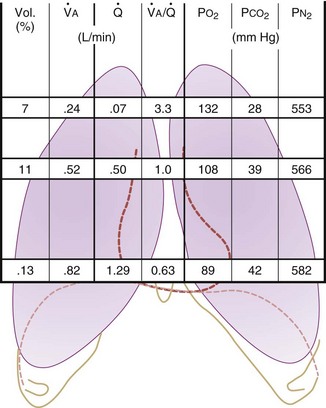
Figure 5-5 Ventilation-perfusion ratio ( ) and the regional composition of alveolar gas. Values for regional flow (
) and the regional composition of alveolar gas. Values for regional flow (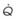 ), ventilation (
), ventilation (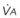 ), partial pressure of oxygen (PO2), and partial pressure of carbon dioxide (PCO2) were derived from Figure 5-4. Partial pressure of nitrogen (PN2) represents what remains from total gas pressure (760 mm Hg including water vapor, which equals 47 mm Hg). The percentage volumes (Vol.) of the three lung slices are also shown. When compared with the top of the lung, the bottom of the lung has a low
), partial pressure of oxygen (PO2), and partial pressure of carbon dioxide (PCO2) were derived from Figure 5-4. Partial pressure of nitrogen (PN2) represents what remains from total gas pressure (760 mm Hg including water vapor, which equals 47 mm Hg). The percentage volumes (Vol.) of the three lung slices are also shown. When compared with the top of the lung, the bottom of the lung has a low  ratio and is relatively hypoxic and hypercapnic.
ratio and is relatively hypoxic and hypercapnic.
(Redrawn from West JB: Regional differences in gas exchange in the lung of erect man. J Appl Physiol 17:893, 1962.)
 inequalities have different effects on arterial CO2 tension (PaCO2) than on arterial O2 tension (PaO2). Blood passing through underventilated alveoli tends to retain its CO2 and does not take up enough O2; blood traversing overventilated alveoli gives off an excessive amount of CO2 but cannot take up a proportionately increased amount of O2 because of the flatness of the oxygen-hemoglobin (oxy-Hb) dissociation curve in this region (see Fig. 5-25). A lung with uneven
inequalities have different effects on arterial CO2 tension (PaCO2) than on arterial O2 tension (PaO2). Blood passing through underventilated alveoli tends to retain its CO2 and does not take up enough O2; blood traversing overventilated alveoli gives off an excessive amount of CO2 but cannot take up a proportionately increased amount of O2 because of the flatness of the oxygen-hemoglobin (oxy-Hb) dissociation curve in this region (see Fig. 5-25). A lung with uneven 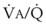 relationships can eliminate CO2 from the overventilated alveoli to compensate for the underventilated alveoli. As a result, with uneven
relationships can eliminate CO2 from the overventilated alveoli to compensate for the underventilated alveoli. As a result, with uneven 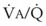 relationships, PACO2-to-PaCO2 gradients are small, and PAO2-to-PAO2 gradients are usually large.
relationships, PACO2-to-PaCO2 gradients are small, and PAO2-to-PAO2 gradients are usually large.
In 1974, Wagner and colleagues described a method of determining the continuous distribution of 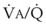 ratios within the lung based on the pattern of elimination of a series of intravenously infused inert gases.16 Gases of differing solubility are dissolved in physiologic saline solution and infused into a peripheral vein until a steady state is achieved (20 minutes). Toward the end of the infusion period, samples of arterial and mixed expired gas are collected, and total ventilation and total cardiac output (
ratios within the lung based on the pattern of elimination of a series of intravenously infused inert gases.16 Gases of differing solubility are dissolved in physiologic saline solution and infused into a peripheral vein until a steady state is achieved (20 minutes). Toward the end of the infusion period, samples of arterial and mixed expired gas are collected, and total ventilation and total cardiac output (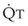 ) are measured. For each gas, the ratio of arterial to mixed venous concentration (retention) and the ratio of expired to mixed venous concentration (excretion) are calculated, and retention-solubility and excretion-solubility curves are drawn. The retention- and excretion-solubility curves can be regarded as fingerprints of the particular distribution of
) are measured. For each gas, the ratio of arterial to mixed venous concentration (retention) and the ratio of expired to mixed venous concentration (excretion) are calculated, and retention-solubility and excretion-solubility curves are drawn. The retention- and excretion-solubility curves can be regarded as fingerprints of the particular distribution of 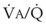 ratios that give rise to them.
ratios that give rise to them.
Figure 5-6 shows the types of distributions found in young, healthy subjects breathing air in the semirecumbent position.17 The distributions of both ventilation and blood flow are relatively narrow. The upper and lower 9% limits shown (vertical interrupted lines) correspond to 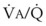 ratios of 0.3 and 2.1, respectively. Note that these young, healthy subjects had no blood flow perfusing areas with very low
ratios of 0.3 and 2.1, respectively. Note that these young, healthy subjects had no blood flow perfusing areas with very low 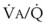 ratios, nor did they have any blood flow to unventilated or shunted areas (
ratios, nor did they have any blood flow to unventilated or shunted areas (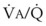 = 0) or unperfused areas (
= 0) or unperfused areas (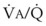 = 8). Figure 5-6 also shows PAO2 and PACO2 in respiratory units with different
= 8). Figure 5-6 also shows PAO2 and PACO2 in respiratory units with different 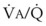 ratios. Within the 95% range of
ratios. Within the 95% range of 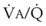 ratios (i.e., 0.3 to 2.1), PO2 ranges from 60 to 123 mm Hg, whereas the corresponding PCO2 range is 44 to 33 mm Hg.
ratios (i.e., 0.3 to 2.1), PO2 ranges from 60 to 123 mm Hg, whereas the corresponding PCO2 range is 44 to 33 mm Hg.
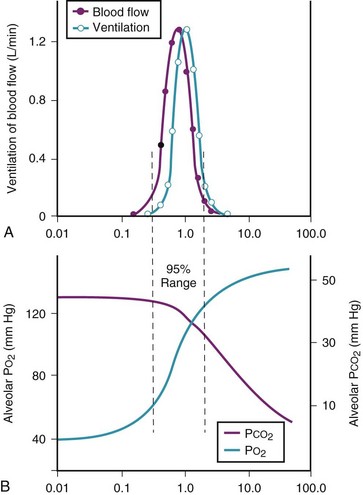
 ) in normal, young, semirecumbent subjects. The 95% range (between dashed lines) is 0.3 to 2.1. B, Corresponding variations in partial pressures of oxygen (P
) in normal, young, semirecumbent subjects. The 95% range (between dashed lines) is 0.3 to 2.1. B, Corresponding variations in partial pressures of oxygen (PB Nongravitational Determinants of Blood Flow Distribution
1 Passive Processes
a Cardiac Output
The pulmonary vascular bed is a high-flow, low-pressure system in health. As 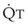 increases, pulmonary vascular pressures increase minimally.18 However, increases in
increases, pulmonary vascular pressures increase minimally.18 However, increases in 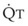 distend open vessels and recruit previously closed vessels. Accordingly, pulmonary vascular resistance (PVR) drops because the normal pulmonary vasculature is quite distensible (and partly because of the addition of previously unused vessels to the pulmonary circulation). As a result of the distensibility of the normal pulmonary circulation, an increase in Ppa increases the radius of the pulmonary vessels, which causes PVR to decrease (Fig. 5-7). Conversely, the opposite effect occurs within the pulmonary vessels during a decrease in
distend open vessels and recruit previously closed vessels. Accordingly, pulmonary vascular resistance (PVR) drops because the normal pulmonary vasculature is quite distensible (and partly because of the addition of previously unused vessels to the pulmonary circulation). As a result of the distensibility of the normal pulmonary circulation, an increase in Ppa increases the radius of the pulmonary vessels, which causes PVR to decrease (Fig. 5-7). Conversely, the opposite effect occurs within the pulmonary vessels during a decrease in 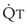 . As
. As 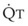 decreases, pulmonary vascular pressures decrease, the radii of the pulmonary vessels are reduced, and PVR consequently increases. The pulmonary vessels of patients with significant pulmonary hypertension are less distensible and act more like rigid pipes. In this setting, Ppa increases much more sharply with any increase in
decreases, pulmonary vascular pressures decrease, the radii of the pulmonary vessels are reduced, and PVR consequently increases. The pulmonary vessels of patients with significant pulmonary hypertension are less distensible and act more like rigid pipes. In this setting, Ppa increases much more sharply with any increase in 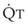 because PVR in these stiff vessels does not decrease significantly due to minimal expansion of their radii.
because PVR in these stiff vessels does not decrease significantly due to minimal expansion of their radii.
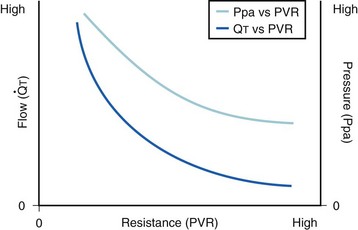
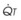 ): PVR = Ppa/
): PVR = Ppa/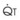 . As
. As 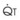 increases, Ppa also increases, but to a lesser extent, and PVR decreases. As
increases, Ppa also increases, but to a lesser extent, and PVR decreases. As 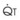 decreases, Ppa also decreases, but to a lesser extent, and PVR increases.
decreases, Ppa also decreases, but to a lesser extent, and PVR increases.Understanding the relationships among Ppa, PVR, and 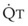 during passive events is a prerequisite to recognition of active vasomotion in the pulmonary circulation (see Lung Volume). Active vasoconstriction occurs whenever
during passive events is a prerequisite to recognition of active vasomotion in the pulmonary circulation (see Lung Volume). Active vasoconstriction occurs whenever 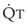 decreases and Ppa either remains constant or increases. Increased Ppa and PVR have been found to be “a universal feature of acute respiratory failure.”19
decreases and Ppa either remains constant or increases. Increased Ppa and PVR have been found to be “a universal feature of acute respiratory failure.”19
 increases and Ppa either remains constant or decreases. When deliberate hypotension is achieved with sodium nitroprusside,
increases and Ppa either remains constant or decreases. When deliberate hypotension is achieved with sodium nitroprusside,  often remains constant or increases, but Ppa decreases, and therefore so does PVR.
often remains constant or increases, but Ppa decreases, and therefore so does PVR.b Lung Volume
Lung volume and PVR have an asymmetric, U-shaped relationship because of the varying effect of lung volume on small intra-alveolar and large extra-alveolar vessels, which in both cases is minimal at functional residual capacity (FRC). FRC is defined as the amount of volume (gas) in the lungs at end-exhalation during normal tidal breathing. Ideally, this means that the patient is inspiring a normal VT, with minimal or no muscle activity or pressure difference between the alveoli and atmosphere at end-exhalation. Total PVR is increased when lung volume is either increased or decreased from FRC (Fig. 5-8).20–22 The increase in total PVR above FRC results from alveolar compression of small intra-alveolar vessels, which results in an increase in small-vessel PVR (i.e., creation of zone 1 or zone 2).23 As a relatively small mitigating or counterbalancing effect to the compression of small vessels, the large extra-alveolar vessels can be expanded by the increased tethering of interstitial connective tissue at high lung volumes (and with spontaneous ventilation only—the negativity of perivascular pressure at high lung volumes). The increase in total PVR below FRC results from an increase in the PVR of large extra-alveolar vessels (passive effect). The increase in large-vessel PVR is partly due to mechanical tortuosity or kinking of these vessels (passive effect). In addition, small or grossly atelectatic lungs become hypoxic, and it has been shown that the increased large-vessel PVR in these lungs is also caused by an active vasoconstrictive mechanism known as hypoxic pulmonary vasoconstriction (HPV).24 The effect of HPV (discussed in greater detail in “Alveolar Gases”) is significant whether the chest is open or closed and whether ventilation is by positive pressure or spontaneous.25
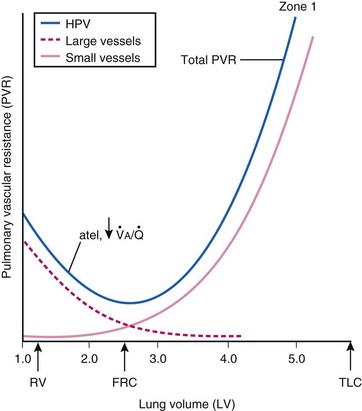
 ) and atelectatic (atel) areas that demonstrate hypoxic pulmonary vasoconstriction (HPV).
) and atelectatic (atel) areas that demonstrate hypoxic pulmonary vasoconstriction (HPV).2 Active Processes and Pulmonary Vascular Tone
Four major categories of active processes affect the pulmonary vascular tone of normal patients: (1) local tissue (endothelial- and smooth muscle–derived) autocrine or paracrine products, which act on smooth muscle (Table 5-1); (2) alveolar gas concentrations (chiefly hypoxia), which also act on smooth muscle; (3) neural influences; and (4) humoral (or hormonal) effects of circulating products within the pulmonary capillary bed. The neural and humoral effects work by means of either receptor-mediated mechanisms involving the autocrine/paracrine molecules listed in Table 5-1 or related mechanisms ultimately affecting the smooth muscle cell.26 These four interrelated systems, each affecting pulmonary vascular tone, are briefly reviewed in sequence.
TABLE 5-1 Local Tissue (Autocrine/Paracrine) Molecules Involved in Active Control of Pulmonary Vascular Tone
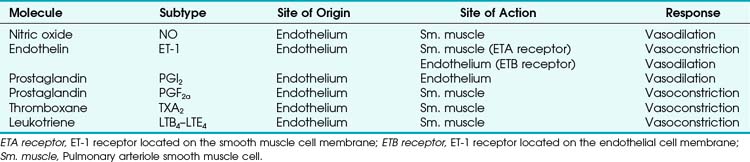
a Tissue (Endothelial- and Smooth Muscle–derived) Products
The pulmonary vascular endothelium synthesizes, metabolizes, and converts a multitude of vasoactive mediators and plays a central role in the regulation of PVR. However, the main effecter site of pulmonary vascular tone is the pulmonary vascular smooth muscle cell (which both senses and produces multiple pulmonary vasoactive compounds).27 The autocrine/paracrine molecules listed in Table 5-1 are all actively involved in the regulation of pulmonary vascular tone during various conditions. Numerous additional compounds bind to receptors on the endothelial or smooth muscle cell membranes and modulate the levels (and effects) of these vasoactive molecules.
Nitric oxide (NO) is the predominant endogenous vasodilatory compound. Its discovery by Palmer and colleagues 25 years ago ended the long search for the so-called endothelium-derived relaxant factor (EDRF).28 Since then, a massive amount of laboratory and clinical research has demonstrated the ubiquitous nature of NO and its predominant role in vasodilation of both pulmonary and systemic blood vessels.29 In the pulmonary endothelial cell, L-arginine is converted to L-citrulline by means of nitric oxide synthase (NOS) to produce the small, yet highly reactive NO molecule.30 Because of its small size, NO can diffuse freely across membranes into the smooth muscle cell, where it binds to the heme moiety of guanylate cyclase (which converts guanosine triphosphate to cyclic guanosine monophosphate [cGMP]).31 cGMP activates protein kinase G, which dephosphorylates the myosin light chains of pulmonary vascular smooth muscle cells and thereby causes vasodilation.31 NOS exists in two forms: constitutive (cNOS) and inducible (iNOS). cNOS is permanently expressed in some cells, including pulmonary vascular endothelial cells, and produces short bursts of NO in response to changing levels of calcium and calmodulin and shear stress. The cNOS enzyme is also stimulated by linked membrane-based receptors that bind numerous molecules in the blood (e.g., acetylcholine, bradykinin).31 In contrast, iNOS is usually produced only as a result of inflammatory mediators and cytokines and, when stimulated, produces large quantities of NO for an extended duration.31 It is well known that NO is constitutively produced in normal lungs and contributes to the maintenance of low PVR.32,33
Endothelin-1 (ET-1) is a pulmonary vasoconstrictor.34 The endothelins are 21-amino-acid peptides that are produced by a variety of cells. ET-1 is the only family member produced in pulmonary endothelial cells, and it is also produced in vascular smooth muscle cells.34 ET-1 exerts its major vascular effects through activation of two distinct G protein–coupled receptors (ETA and ETB). ETA receptors are found in the medial smooth muscle layers of the pulmonary (and systemic) blood vessels and in atrial and ventricular myocardium.34 When stimulated, ETA receptors induce vasoconstriction and cellular proliferation by increasing intracellular calcium.35 ETB receptors are localized on endothelial cells and some smooth muscle cells.36 Activation of ETB receptors stimulates the release of NO and prostacyclin, thereby promoting pulmonary vasodilation and inhibiting apoptosis.30 Bosentan, an ET-1 receptor antagonist, has produced modest improvement in the treatment of pulmonary hypertension.37 The more selective ETA receptor antagonist, sitaxsentan, showed additional benefit in improving pulmonary hypertension.38 However, both of these ET-1 receptor antagonists are associated with an increased risk of liver toxicity.39 In summary, it appears that there is a normal balance between NO and ET-1, with a slight predominance toward NO production and vasodilation in health.
Similarly, various eicosanoids are elaborated by the pulmonary vascular endothelium, with a balance toward the vasodilatory compounds in health. Prostaglandin I2 (PGI2), now known as epoprostenol (previously known as prostacyclin), causes vasodilation and is continuously elaborated in small amounts in healthy endothelium. In contrast, thromboxane A2 and leukotriene B4 are elaborated under pathologic conditions and are thought to be involved in the pathophysiology of pulmonary artery hypertension (PAH) associated with sepsis and reperfusion injury.26
Therapeutically, epoprostenol has been used successfully to decrease PVR in patients with chronic PAH when infused or inhaled.40,41 Currently, the synthetic PGI2 (iloprost) is the most commonly used inhaled eicosanoid for reduction of PVR in patients with PAH.41Although most patients with chronic PAH are unresponsive to an acute vasodilator challenge with short-acting agents such as epoprostenol, adenosine, or NO,42 long-term administration of epoprostenol has been shown to decrease PVR in these patients.43 Furthermore, some patients with previously severe PAH have been weaned from epoprostenol after long-term administration, with dramatically decreased PVR and improved exercise tolerance.42 The vascular remodeling required to provide such a dramatic reduction in PVR is probably the result of mechanisms besides simple local vasodilation, as predicted by Fishman in an editorial in 1998.44 One such mechanism that appears to be important is the increased clearance of ET-1 (a potent vasoconstrictor and mitogen) with long-term epoprostenol administration.45
b Alveolar Gases
Hypoxia-induced vasoconstriction constitutes a fundamental difference between pulmonary vessels and all other systemic blood vessels (which vasodilate in the presence of hypoxia). Alveolar hypoxia of in vivo and in vitro whole lung, unilateral lung, lobe, or lobule of lung results in localized pulmonary vasoconstriction. This phenomenon is widely referred to as HPV and was first described more than 65 years ago by Von Euler and Liljestrand.46 The HPV response is present in all mammalian species and serves as an adaptive mechanism for diverting blood flow away from poorly ventilated to better ventilated regions of the lung and thereby improving 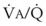 ratios.47 The HPV response is also critical for fetal development because it minimizes perfusion of the unventilated lung.
ratios.47 The HPV response is also critical for fetal development because it minimizes perfusion of the unventilated lung.
The HPV response occurs primarily in pulmonary arterioles of about 200 µm internal diameter (ID) in humans (60 to 700 µm ID in other species).48 These vessels are advantageously situated anatomically in close relation to small bronchioles and alveoli, which permits rapid and direct detection of alveolar hypoxia. Indeed, blood may actually become oxygenated in small pulmonary arteries because of the ability of O2 to diffuse directly across the small distance between the contiguous air spaces and vessels.49 This direct access that gas in the airways has to small arteries makes possible a rapid and localized vascular response to changes in gas composition.
The O2 tension at the HPV stimulus site (PsO2) is a function of both PAO2 and mixed venous O2 pressure (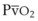 ).50 The PsO2-HPV response is sigmoid, with a 50% response when PAO2,
).50 The PsO2-HPV response is sigmoid, with a 50% response when PAO2, 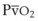 , and PsO2 are approximately 30 mm Hg. Usually, PAO2 has a much greater effect than
, and PsO2 are approximately 30 mm Hg. Usually, PAO2 has a much greater effect than 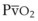 does because O2 uptake is from the alveolar space to the blood in the small pulmonary arteries.50
does because O2 uptake is from the alveolar space to the blood in the small pulmonary arteries.50
Numerous theories have been developed to explain the mechanism of HPV.46,51–53 Many vasoactive substances have been proposed as mediators of HPV, including leukotrienes, prostaglandins, catecholamines, serotonin, histamine, angiotensin, bradykinin, and ET-1, but none has been identified as the primary mediator. In 1992, Xuan proposed that NO has a pivotal role in modulating PVR.54 NO is involved, but not precisely in the way that Xuan first proposed. There are multiple sites of O2 sensing with variable contributions from the NO, ET-1, and eicosanoid systems (described earlier). In vivo, HPV is currently thought to result from the synergistic action of molecules produced in both endothelial cells and smooth muscle cells.55 However, HPV can proceed in the absence of intact endothelium, suggesting that the primary O2 sensor is in the smooth muscle cell and that endothelium-derived molecules modulate only the primary HPV response.
The precise mechanism of HPV is still under investigation. However, current data support a mechanism involving the smooth muscle mitochondrial electron transport chain as the HPV sensor (Fig. 5-9).56 In addition, reactive oxygen species (possibly H2O2 or superoxide) are released from complex III of the electron transport chain and probably serve as second messengers to increase calcium in pulmonary artery smooth muscle cells during acute hypoxia.57 However, alternative (less likely) mechanisms are still being investigated.58 One alternative hypothesis suggests that smooth muscle microsomal reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidoreductase or sarcolemmal NADPH oxidase is the sensing mechanism.58 Another, previously popular theory posited that voltage-sensitive potassium (KV) channels were required for the HPV response. However, KV channels are no longer believed to be obligate but instead are thought to be attenuators, because a study demonstrated that inhibition of KV channels failed to inhibit the HPV response.58
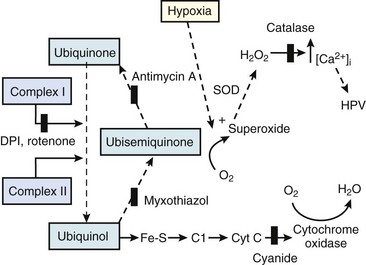
The clinical effects of HPV in humans can be classified under three basic mechanisms. First, life at high altitude or whole-lung respiration of a low inspired concentration of O2 (FIO2) increases Ppa. This is true for newcomers to high altitude, for the acclimatized, and for natives.53 The vasoconstriction is considerable; in healthy people breathing 10% O2, Ppa doubles whereas pulmonary wedge pressure remains constant.59 The increased Ppa increases perfusion of the apices of the lung (through recruitment of previously unused vessels), which results in gas exchange in a region of lung not normally used (i.e., zone 1). Therefore, with a low FIO2, PAO2 is greater and the alveolar-arterial O2 tension difference and the ratio between dead space and tidal volume (VD/VT) are less than would be expected or predicted on the basis of a normal (sea level) distribution of ventilation and blood flow. High-altitude pulmonary hypertension is an important component in the development of mountain sickness subacutely (hours to days) and cor pulmonale chronically (weeks to years).60 There is now good evidence that in both patients with chronic obstructive pulmonary disease (COPD) and those with obstructive sleep apnea (OSA), nocturnal episodes of arterial O2 desaturation (caused by episodic hypoventilation) are accompanied by elevations in Ppa that can eventually lead to sustained pulmonary hypertension and cor pulmonale.61
Second, hypoventilation (low 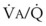 ratio), atelectasis, or nitrogen ventilation of any region of the lung usually causes a diversion of blood flow away from the hypoxic to the nonhypoxic lung (40% to 50% in one lung, 50% to 60% in one lobe, 60% to 70% in one lobule) (Fig. 5-10).62 The regional vasoconstriction and blood flow diversion are of great importance in minimizing transpulmonary shunting and normalizing regional
ratio), atelectasis, or nitrogen ventilation of any region of the lung usually causes a diversion of blood flow away from the hypoxic to the nonhypoxic lung (40% to 50% in one lung, 50% to 60% in one lobe, 60% to 70% in one lobule) (Fig. 5-10).62 The regional vasoconstriction and blood flow diversion are of great importance in minimizing transpulmonary shunting and normalizing regional 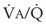 ratios during disease of one lung, one-lung anesthesia (see Chapter 26), inadvertent intubation of a main stem bronchus, and lobar collapse.
ratios during disease of one lung, one-lung anesthesia (see Chapter 26), inadvertent intubation of a main stem bronchus, and lobar collapse.
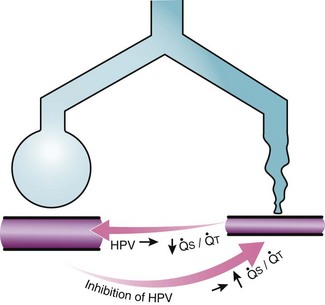
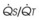 ) that can occur through the hypoxic lung. Inhibition of hypoxic lung HPV causes an increase in the amount of shunt flow through the hypoxic lung, thereby decreasing the alveolar oxygen tension (P
) that can occur through the hypoxic lung. Inhibition of hypoxic lung HPV causes an increase in the amount of shunt flow through the hypoxic lung, thereby decreasing the alveolar oxygen tension (PThird, in patients who have COPD, asthma, pneumonia, or mitral stenosis but not bronchospasm, administration of pulmonary vasodilator drugs such as isoproterenol, sodium nitroprusside, or nitroglycerin inhibits HPV and causes a decrease in PaO2 and PVR and an increase in right-to-left transpulmonary shunting.63 The mechanism for these changes is thought to be deleterious inhibition of preexisting and, in some lesions, geographically widespread HPV without concomitant and beneficial bronchodilation.63 In accordance with the latter two lines of evidence (one-lung or regional hypoxia and vasodilator drug effects on whole-lung or generalized disease), HPV is thought to divert blood flow away from hypoxic regions of the lung, thereby serving as an autoregulatory mechanism that protects PaO2 by favorably adjusting regional 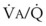 ratios. Factors that inhibit regional HPV are extensively discussed elsewhere.64,65
ratios. Factors that inhibit regional HPV are extensively discussed elsewhere.64,65
c Neural Influences on Pulmonary Vascular Tone
The three systems used to innervate the pulmonary circulation are the same ones that innervate the airways: the sympathetic, parasympathetic, and nonadrenergic noncholinergic (NANC) systems.26 Sympathetic (adrenergic) fibers originate from the first five thoracic nerves and enter the pulmonary vessels as branches from the cervical ganglia, as well as from a plexus of nerves arising from the trachea and main stem bronchi. These nerves act mainly on pulmonary arteries down to a diameter of 60 µm.26 Sympathetic fibers cause pulmonary vasoconstriction through α1-receptors. However, the pulmonary arteries also contain vasodilatory α2-receptors and β2-receptors. The α1-adrenergic response predominates during sympathetic stimulation, such as occurs with pain, fear, and anxiety.26 The parasympathetic (cholinergic) nerve fibers originate from the vagus nerve and cause pulmonary vasodilation through an NO-dependent process.26 Binding of acetylcholine to a muscarinic (M3) receptor on the endothelial cell increases intracellular calcium and stimulates cNOS.26 NANC nerves cause pulmonary vasodilation through NO-mediated systems by using vasoactive intestinal peptide as the neurotransmitter. The functional significance of this system is still under investigation.26
Related posts:
 Prehospital Airway Management
Prehospital Airway Management
 Medical-Legal Considerations: The ASA Closed Claims Project
Medical-Legal Considerations: The ASA Closed Claims Project
 Performance of Rigid Bronchoscopy
Performance of Rigid Bronchoscopy
 Intubating Introducers, Stylets, and Lighted Stylets (Lightwands)
Intubating Introducers, Stylets, and Lighted Stylets (Lightwands)
 Percutaneous Dilational Cricothyrotomy and Tracheostomy
Percutaneous Dilational Cricothyrotomy and Tracheostomy

Full access? Get Clinical Tree


