Chapter 25 Pharmacology of the Cardiovascular System
Mechanisms of Response
Adrenergic Receptors
Catecholamines modify cellular physiology by interacting with a specific adrenergic receptor. The classic paradigm of α and β classes of adrenergic receptors remains unchanged, although new subtypes and sub-subtypes continue to be investigated. Currently three subtypes of α1 receptors and three subtypes of α2 receptors have been described and three subtypes of β receptors are recognized.1 Advances in the biology of the adrenergic receptor have led to a greater understanding of role of the α receptor in the heart, adrenergic receptor regulation of cardiac myocyte apoptosis, and the coupling of the β2 receptor to more than one G protein. The discovery of various polymorphisms for the adrenergic receptors has served to add even more complexity, but the clinical relevance of many of these polymorphisms has yet to be elucidated. Despite this increase in our understanding of the adrenergic receptor, the clinical classification of the catecholamines into α and β agents remains functionally unchanged (Table 25-1).
Table 25–1 Adrenergic Receptors: Physiologic Responses, Agonist Potency, and Representative Antagonists
Signal Transduction
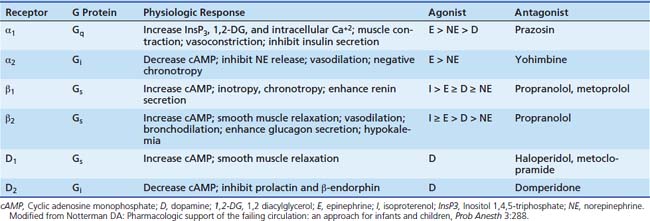
Adrenergic receptors mediate their effects through G proteins and as such are classified as G protein coupled receptors. The adrenergic receptor itself contains seven membrane-spanning α-helical domains, an extracellular N-terminal segment, and a cytosolic C-terminal segment (Figure 25-1). G proteins are heterotrimeric proteins consisting of α, β, and γ subunits, each of which has multiple subfamilies.2 There are at least 20 α subunits, five β subunits, and six γ subunits. The action resulting from a ligand binding to a particular adrenergic receptor is a function of the specific type of subunits compromising the G protein receptor complex. An evolving concept is spontaneous receptor activation, as described for the β2 adrenergic receptor.3,4 In this model, the receptor “toggles” between different conformational states, of which some are active and each of which may be bound to different G proteins. Binding of a ligand may stabilize or invoke a shift to a particular conformation and in doing so promote changes in the expression of second messengers, most commonly protein kinases. Hence the response of ligand binding may not only depend on the ligand involved but the state of the receptor at the time of binding.
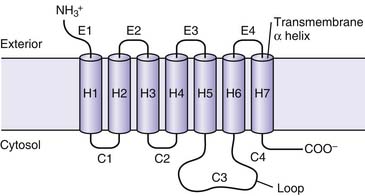
Adrenergic receptors typically are coupled to one of three types of G proteins: Gs, Gi, or Gq. Gs proteins produce an increase in adenylate cyclase activity, while Gi proteins promote a decrease in adenylate cyclase activity. Gq protein receptors stimulate phospholipase C to generate diacylglycerol and inositol 1,4,5-triphosphate. The nature of the G protein is usually a function of the type of α subunit (αs, αI, αq, and α12/13).5 Events involving interaction of G proteins, the receptor protein, and adenylate cyclase are summarized in Figure 25-2. In the example of the Gs protein, ligand binding to the coupled receptor causes a conformational change in the G protein, resulting in guanosine diphosphate (GDP) disassociating from the Gsα subunit and guanosine triphosphate (GTP) binding to the α subunit. This GTP-Gα complex then disassociates from the Gβγ subunit and binds to adenylate cyclase, leading to an increase in activity of this enzyme. Adenylate cyclase catalyses the conversion of adenosine triphosphate (ATP) to cyclic adenosine monophosphate (cAMP), thus increasing cellular levels of cAMP. Gi proteins have a different α subunit; when the Giα GTP complex binds to adenylate cyclase, the enzyme is inactivated. By inhibiting this enzyme, Gi-coupled receptor agonists produce a decrease in the cellular concentration of cAMP. The specific cellular response that follows an alteration in the concentration of cAMP depends on the specialized function of the target cell.6 Typically, an increase in concentration of cAMP leads to activation of a cAMP-dependent protein kinase. These kinases then phosphorylate and activate other structures and enzymes. Many compounds other than adrenergic agents also increase intracellular levels of cAMP. The question of how different agents produce specific responses through the expression of common second messengers continues to be investigated. One proposed mechanism involves anchoring proteins, such as A kinase anchoring proteins. These proteins localize protein kinase A (PKA) to particular cellular locales and also may offer binding sites for other regulatory proteins.7 Similarly, anchoring proteins for both the active and inactive forms of protein kinase C have been described.8 Different subtypes of anchoring proteins may serve to create another level of specificity in the effector response for a particular ligand by confining the response to a particular area.
β-Adrenergic Receptors
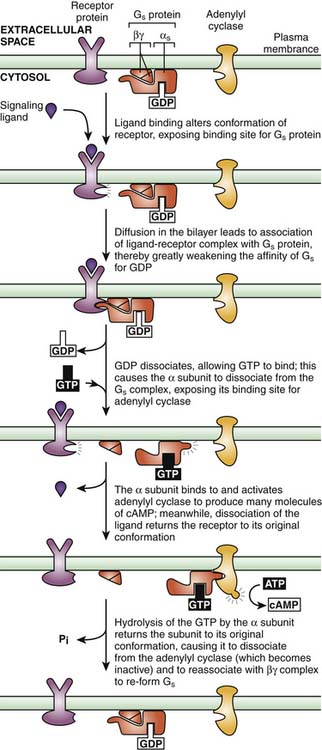
Myocardial β1-adrenergic receptors are associated with Gs. When this receptor type is engaged by an agonist agent, the result is enhanced activity of adenylate cyclase and a rise in the concentration of cAMP. This process activates PKA. PKA in turn phosphorylates voltage-dependent calcium channels, increasing the fraction of channels that can be open and the probability that these channels are open, producing an increase in intracellular calcium concentration (Figure 25-3).9 Calcium then binds to troponin C, allowing for actin-myosin cross bridge formation and sarcomere contraction. In addition, PKA phosphorylates phospholamban, relieving the disinhibitory effect of the unphosphorylated form on calcium channels in the sarcoplasmic reticulum. The accumulation of calcium by the sarcoplasmic reticulum is thus enhanced, increasing the rate of sarcomere relaxation (lusitropy) and subsequently increasing the amount of calcium available for the next contraction. This process leads to both enhanced contractility and active diastolic relaxation.
The question of a PKA-independent mechanism by which β-adrenergic receptors can activate calcium channels remains controversial.10 Experimental evidence indicates that β1 receptors are preferentially coupled to cardiac calcium channels in a cAMP-independent mechanism; however, a binding site on the calcium channel has not been delineated.11,12 β2-adrenergic receptors have been shown to physically complex, with the calcium channel v1.2 in neuronal tissue.13 Of note, whereas it was once believed that the β2 receptor had little presence and no role in the heart, recent evidence suggests just the opposite. β2 receptors are present in the heart and in fact couple with both Gs and Gi.14 The response to β2 activation in the heart remains controversial. β2 activation is actually more effective at increasing cAMP levels than β1 activation, but the effect on PKA is localized rather than diffuse, as is the case following β1 activation. This effect could be due to the localization of receptor signaling by the Gi component. Evidence for this hypothesis is that treatment with pertussis toxin (an inhibitor of Gi) transforms the β2 signal from a local one to a diffuse one.15 In summary, the role of β2 receptors in the heart and the signaling processes involved are not fully resolved.5,16 Their role in the failing heart and implications for disease also remain to be elucidated.17,18 To add further complexity, functional β3 receptors have been demonstrated in the heart and appear to have a negative inotropic effect.19
In vascular smooth muscle, both β1 and β2 receptors are present, although β2 receptors predominate.1 The β2 receptor is coupled to Gs; therefore, activation of β2 receptors promotes formation of cAMP. The resulting activation of cAMP-dependent protein kinase in vascular smooth muscle, however, stimulates pumps that remove calcium from the cytosol and also promotes calcium uptake by the sarcoplasmic reticulum. As cytosolic calcium concentration decreases, smooth muscle relaxes and the blood vessel dilates. Adrenergic receptors also have been demonstrated on the endothelium and are capable of producing relaxation of the vessel.20 The exact mechanism involved, including the role of nitric oxide and the subtype of β receptors involved, remains under investigation.1,21
α Receptors
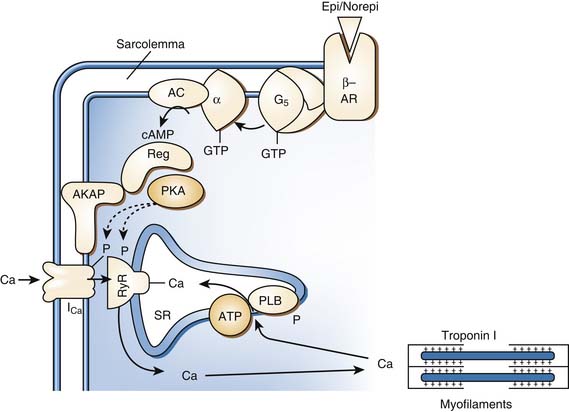
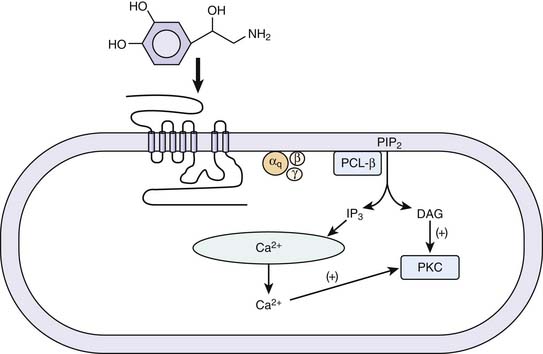
Vascular smooth muscle contraction is mediated via α1 adrenergic receptors, of which there are three subtypes: 1A, 1B, and 1D. The individual contributions of each of these subtypes to the control of vascular tone remains an active area of investigation. Each subtype may be expressed in all of the vascular beds, but it is thought that one type will predominate for a particular bed.22 A mouse knockout model of the α1D receptor showed that α1 binding in the aorta was lost but preserved in the heart. The knockout model also had lower blood pressures and a decreased response to norepinephrine.23 α1A and α1B are thought to be involved in both the heart and vasculature.24,25 A knockout model of α1A demonstrated decreased blood pressure and response to phenylephrine.22 An animal model of overexpression of the α1A receptor was associated with marked increase in cardiac contractility without a change in blood pressure or heart rate.26 In a knockout model of α1B receptors in mice, chronic exposure to norepinephrine did not lead to cardiac hypertrophy or vascular remodeling.27 Overexpression of a mutant α1B receptor led to increased expression of mitogen-activated protein kinases and decreased responsiveness to isoproterenol in isolated cardiac preparations.28 Thus while α receptors may have an inotropic effect less than that of β-adrenergic receptors, they do have significant effects in the myocardium. Interestingly, in patients with heart failure, downregulation of β receptors has been noted while α receptors are preserved.29 The α1 receptor is coupled to the family of Gq/11 proteins, which act independently of cAMP. Signal transduction across this receptor is initiated by the activation of phospholipase C, which hydrolyzes phosphatidylinositol 4,5-biphosphate to inositol 1,4,5-triphosphate (InsP3) and 1,2 diacylglycerol (1,2DG). InsP3 binds to specific receptors on the sarcoplasmic reticulum, causing a release of calcium into the cytosol, and promotes movement of extracellular calcium into the cell. 1,2DG with calcium activates protein kinase C (PKC), which regulates movement of calcium into the cytosol (Figure 25-4). In vascular smooth muscle, medium light chain kinase is activated as a result and phosphorylates myosin light chain 2, leading to smooth muscle contraction.30 It also has been shown that a similar mechanism underlies the inotropic effect of the α1 receptor in the myocardium.31 The α1A receptor appears to be the most efficiently coupled of the different subtypes.32 The α1 receptors also activate calcium influx through voltage-dependent and voltage-independent calcium channels.33 The α receptors also promote the activation of the mitogen-activated kinase family, which are key regulators of cell growth.
Receptor Downregulation
The mechanisms described provide numerous sites at which the activity of the system can be modified, thereby affecting the sensitivity of target cells to both exogenous and endogenous catecholamines. Some of these receptor modifications are clinically important to the critical care physician. The best-documented type of modification involves agonist-mediated receptor desensitization. Exposure of receptors to agonists markedly reduces the sensitivity of the target cell to the agonist. Within seconds to minutes after agonist binding, the receptor may be uncoupled as a result of receptor phosphorylation. The receptor may be phosphorylated by PKA or PKC or by a member of the family of G receptor kinases (GRKS). These kinases, which include β-adrenergic receptor kinases 1 and 2, phosphorylate only receptors that have agonist bound. Compared with PKA and PKC, phosphorylation by GRKS enhances the ability of β arrestin, a cytosolic protein, or clathrin to bind to the receptor and disrupt further signaling. The role of GRKS has been established for β1, β2, and α2 receptors.17,34 Sequestration of receptors within the target cell and degradation of sequestered receptors is another mechanism by which receptors are downregulated. The desensitization of α1 receptors has been extensively reviewed.35 Homologous desensitization is mediated by GRKS, which are activated by soluble Gβγ subunits and phosphatidylinositol biphosphate. As with the other adrenergic receptors, once phosphorylated, the receptors are internalized into vesicles. The α1 receptors also demonstrate heterologous desensitization, in which a second messenger kinase, generated as a result of ligand binding, inactivates the receptor and prevents any further signaling from the receptor. In addition to agonist-mediated desensitization, other stimuli also have been implicated in downregulation, including endotoxin, tumor necrosis factor, and congestive heart failure (CHF).36 Lymphocyte β-adrenergic receptor density in children with CHF was reduced in proportion to the degree of elevation in plasma norepinephrine concentration.37 Several pharmacologic agents, such as corticosteroids and ketotifen, are thought to upregulate β-adrenergic receptors, and evidence exists that both immaturity and senescence are associated with β-adrenergic receptor desensitization.38
Polymorphisms
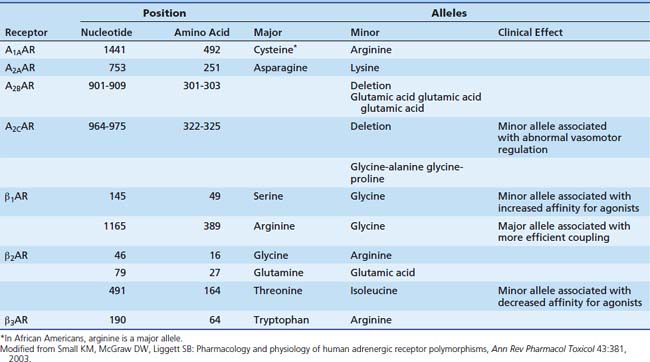
With the rapid advances in molecular biology, knowledge of polymorphisms within the genetic code has increased dramatically. Polymorphisms within the adrenergic receptors (see Table 25-2) is reviewed briefly here. The reader also is directed toward a recent comprehensive review.39 Two polymorphisms have been described in the β1 receptor. At position 49, glycine may be substituted for serine. The frequency of the minor allele (gly) is about 15%. In cell culture studies, the minor allele is associated with enhanced agonist-promoted downregulation and increased affinity for agonists.40 Another polymorphism occurs at position 389, where glycine may be substituted for arginine. This position is in the carboxy terminus, a site involved with binding with the G protein. In a fibroblast cell line, the arginine alleles (wild type) were observed to have higher levels of adenyl cyclase expression with agonist binding, representing more efficient coupling.41 In a model utilizing isolated human myocardial tissue, the wild type allele (Arg 389) was associated with enhanced inotropic potency in response to norepinephrine, although maximal force generated did not differ between the two alleles.42 Another study demonstrated higher resting heart rates and diastolic blood pressure in patients with the Arg 389 allele.43 Three polymorphisms have been demonstrated in the β2 receptor. Of note, at position 164, isoleucine may be substituted for threonine. This polymorphism is associated with a threefold decrease in affinity for agonist binding.44 In a transfected cell line, the Ile-164 type is associated with decreased basal adenyl cyclase activity and decreased activity after agonist stimulation.45 Similar results were found in a study of patients heterozygous for the Ile-164 allele. These patients had blunting of the increase in both heart rate and duration of systole during terbutaline infusion, with a trend toward lower systolic blood pressure.46 A single polymorphism has been delineated in the α1 receptor; it does not appear to have any clinical significance. A restriction fragment length polymorphism was identified in the gene for the α2C receptor.47 This polymorphism was associated with abnormal vasomotor regulation, sodium excretion, and platelet function. The presence of both the Arg 389 β1 receptor polymorphism and a deletion polymorphism in the α2C receptor in black patients has been shown to increase the risk of heart failure.48 This study is an example of the difficulty in linking genetic variations to the clinical scenario in that a single polymorphism may not have an effect except in the presence of another polymorphism. None of these particular polymorphisms has yet been shown to have a significant role in the pediatric intensive care unit (PICU), but they no doubt will continue to add to our understanding of the adrenergic pathways.
Vasopressin Receptors
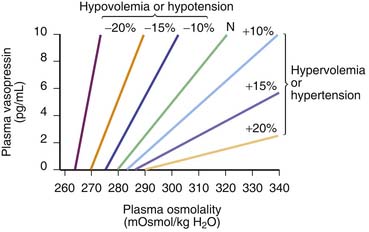
Arginine vasopressin (AVP) is a nonpeptide hormone synthesized in the supraoptic and paraventricular nuclei of the hypothalamus. Three subtypes of vasopressin receptors exist, known as V1, V2, and V3 (or V1b). V2 receptors are present in the renal collecting duct, while V1 receptors are located in the vascular bed, kidney, bladder, spleen, and hepatocytes, among other tissues.49 AVP is released in response to small increases in plasma osmolality or large decreases in blood pressure or blood volume.50 The plasma osmolality threshold for release of AVP is 280 mOsm/kg; above this threshold there is a steep linear relation between serum osmolality and AVP levels.50 Changes of at least 20% in blood volume are needed to effect a change in AVP levels, although levels may then increase by twentyfold to thirtyfold.50 Hypovolemia also shifts the response curve for AVP to osmolalar changes to the left and increases the slope of the curve (Figure 25-5). Vasopressin can produce vasoconstriction through V1 receptors in the vascular bed (discussed later), but it also activates V1 receptors in the central nervous system (CNS), including receptors in the area postrema.51 This region is responsible for the reflex bradycardia seen with AVP infusion. This reflex attenuates the increase in blood pressure that would result from the vasoconstrictor effects of AVP.52,53 In fact, vasopressin causes a greater reduction in heart rate than other vasoconstrictors.49 Thus if this feedback loop is abolished, AVP induces a greater vasopressor response than other agents.51
V1 Receptors
Vasopressin receptors belong to the family of G protein–coupled receptors. V1 receptors are coupled to Gq and V2 receptors are coupled to Gs.50 When vasopressin binds to the V1 receptor, phospholipase C is activated with the eventual production of InsP3 and 1,2 DG. These molecules serve to increase the release of calcium from the endoplasmic reticulum as well as increase the entry of calcium through gated channels (Figure 25-6).54 The increase in intracellular calcium leads to an increase in the activity of myosin light chain kinase. This kinase acts upon myosin to increase the number of actin-myosin cross bridges, enhancing contraction of the myocyte. Of note, vasopressin has been shown to produce vasoconstriction in the skin, skeletal muscle, and fat while producing vasodilatation in the renal, pulmonary, and cerebral vasculature.55 This effect may be mediated though nitric oxide or may be a function of the isoform of adenyl cyclase with which the receptor is coupled.56 AVP also has been shown to increase the pressor effects of catecholamines, although in two different vascular smooth muscle cell lines, AVP had opposing effects on isoproterenol-induced activation of adenyl cyclase.57–59 Other effects of vasopressin binding to V1 receptors are also shown in Figure 25-6.
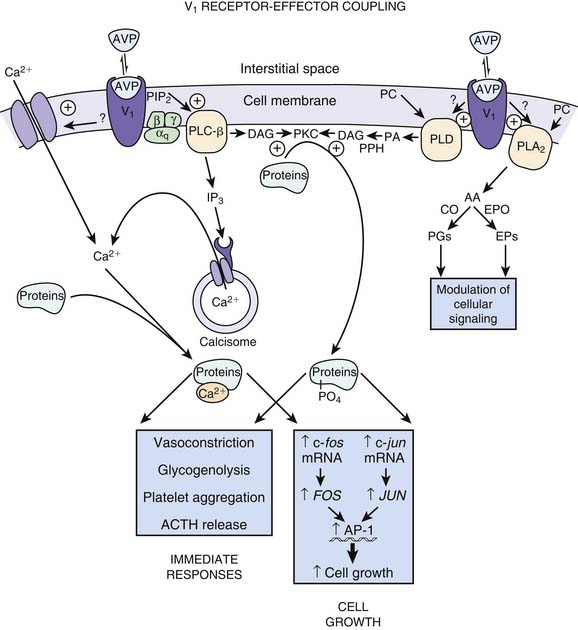
AVP also may increase vascular tone by interacting with so-called ATP-sensitive potassium channels termed KATP.60 Activation of KATP channels hyperpolarizes the cell, closes calcium channels, and prevents contraction.61,62 The result may be to protect the cell.63 AVP can induce PKC, which in turn inhibits the KATP channel when the cellular concentration of ATP is low.64 Inhibition of the KATP channels allows for cell depolarization and calcium entry, resulting in vasoconstriction (Figure 25-7).65 V1 receptors also have been demonstrated to have a weak positive inotropic effect in the heart, although the clinical significance of this effect has not been established.66
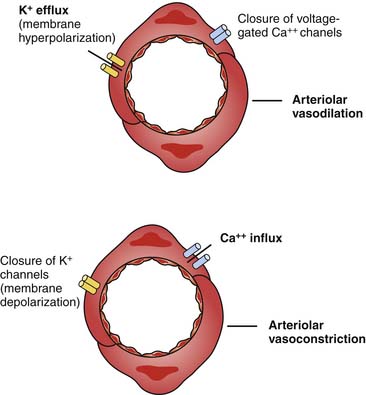
Receptor Downregulation
As with adrenergic receptors, vasopressin receptors undergo downregulation. AVP promotes the phosphorylation of it own receptor immediately after binding. The receptor is removed from the cell surface within 3 minutes after binding.67 As with adrenergic receptors, G protein–coupled receptor kinases catalyze the phosphorylation of the receptor. PKC also mediates this reaction and may serve as the means by which other agents downregulate the vasopressin receptor in a heterologous manner.67
Phosphodiesterase Regulation of cAMP
Phosphodiesterases are a class of enzyme that catalyze the hydrolysis of cAMP and cGMP into AMP and GMP, respectively. Therefore, these enzymes can downregulate the signals transducted by cAMP, such as PKA activity (discussed previously). Several families of this enzyme exist, each with subtypes. Phosphodiesterase III (PDE3) is present in many cell types, including cardiac myocytes, vascular smooth muscle cells, adipocytes, platelets, and pancreatic islet cells. PDE3 has a much higher Vmax for cAMP than it does for cGMP and therefore is functionally a cAMP esterase.69 Different isoforms of PDE3 are present in cardiac (PDE3A1) and vascular smooth muscle cells (PDE3A2) and are localized to different cellular compartments and as such are able to regulate the function of their target enzymes in response to particular cellular signals.70 The C-terminus of the enzyme codes for the central catalytic core that is the active binding site and the N-terminus coding region may determine cellular targeting.70 The bipyridines, such as milrinone, are competitive inhibitors of PDE3; that is, they bind to PDE3, preventing the enzyme from binding to cAMP.71,72 They appear to bind near the binding site for cAMP, although different PDE3 inhibitors may have different binding sites.73 Inhibition of PDE3 produces an increase in cAMP. This increase in cAMP results in a positive inotropic effect in the myocardium and vasodilatation in the systemic and pulmonary vasculature.74 In contrast, methylxanthines such as theophylline, which inhibit all phosphodiesterases, cause levels of both cGMP (thought to decrease contractility) and cAMP to increase. This dual increase attenuates their inotropic effects. Bipyridines also may enhance contractility by increasing the sensitivity of myofilaments to cytosolic calcium.75 Milrinone has also been shown to enhance the sarcomere uptake of calcium and thereby augment left ventricular (LV) relaxation (lusitropy).76 In the peripheral vasculature, PD3 inhibitors may produce vasodilatation via a cGMP mechanism.77 The bipyridines offer the combination of positive inotropy, lusitropy, and afterload reduction. It has been speculated that long-term exposure to PDE inhibitors may result in adrenergic receptor desensitization via heterologous desensitization, although this has not been demonstrated clinically.78
ATPase Inhibition
Membrane-bound sodium-potassium adenosine triphosphatase (Na-K ATPase) is responsible for maintaining electrochemical gradients across the cellular membrane. It does so by extruding three molecules of sodium from the cell and incorporating two molecules of potassium into the cell, both against their respective concentration gradients. This process occurs at the cost of one molecule of ATP. The enzyme consists of an α and β subunit; there are four subtypes of α and three of β.79 The β subunit may be involved in the trafficking of the enzyme.80 The α subunit contains both the binding site and catalytic site.79 The isoforms expressed are dependent on the type of tissue.81 The putative mechanism of action of the cardiac glycosides is inhibition of the Na-K ATPase pump, which results in an increase in intracellular sodium. The elevated level of intracellular sodium then effects a decrease in the activity of the sodium/calcium exchange (NCX) pump. This pump exchanges three molecules of extracellular sodium for one molecule of intracellular calcium.82,83 The net result is a rise in intracellular calcium and, in the cardiac myocyte, enhanced contractility. The particular α isoform expressed may affect the function of the enzyme. In a mouse knockout model, animals heterozygous for α1 (α1−/+) and α2 (α2−/+) were generated.84 The cardiac myocytes from α2 heterozygous animals demonstrated a hypercontractile state when compared with control subjects (similar to that seen with the administration of cardiac glycosides). In contrast, the α1 heterozygous animals had a hypocontractile state. The authors note that this phenotype is similar to that seen with cardiac glycoside toxicity. Although the classical explanation has been described, the precise mechanism for the increase in intracellular calcium with Na-K ATPase inhibition remains controversial. Some studies have demonstrated a rise in intracellular calcium without the expected concomitant rise in intracellular sodium, suggesting that perhaps the NCX pump was not involved.85,86 In contrast, a study in mice lacking the NCX pump demonstrated that ouabain (a Na-K ATPase inhibitor) did not increase intracellular calcium.87,88 No evidence exists to suggest the development of tolerance to digoxin with long-term use.89
Developmental Issues
It is often stated that the immature myocardium is less sensitive to inotropic agents than the adult heart. The exact nature of the mechanism responsible for these differences represent an area of active investigation. The majority of studies involve animal models or isolated human tissue. Because there are inherent differences between intact healthy animal models and the ill child, as well as pharmacokinetic differences between the infant and adult patient, caution must be exercised when extrapolating laboratory data to the bedside.90 Nonetheless, a brief review of developmental differences is appropriate.
Age-related differences exist in the response of the developing myocardium to inotropic agents, receptor regulation, and calcium handling.82,91 In puppy heart models, the inotropic response to dopamine and isoproterenol increases with increasing age.92,93 Maximal developed pressure and relaxation velocity in response to isoproterenol were higher in adult rabbit hearts compared with neonatal hearts.94 However, in a model of rat ventricular myocytes, zinterol (a selective β2 agonist) increased intracellular calcium gradients and cAMP accumulation and augmented cell shortening in neonatal myocytes at much lower concentrations than in adult myocytes.95 Sun,96 in a rabbit model, showed that isolated adult hearts had a greater increase in systolic function in response to isoproterenol than did neonatal hearts but that the response to continuous infusions was less attenuated in the neonatal hearts, suggesting less desensitization. In another study,97 neonatal rat hearts did not show any evidence of homologous uncoupling in response to isoproterenol. In fact, the receptors demonstrated an enhanced response after prolonged exposure to agonist. This response occurred despite increased activity of β-adrenergic receptor kinase 1, which mediates receptor uncoupling and sequestration. The ability of the neonate to resist desensitization may be helpful given the higher levels of catecholamines present in the newborn period. The mechanism by which neonatal receptors accomplish this was examined in a rat model of the response of cardiac and hepatic cells to isoproterenol and terbutaline.98 Neonatal hearts did not show any evidence of desensitization with either single injections of or prolonged exposure to isoproterenol or terbutaline. Adult hearts had evidence of both homologous and heterologous desensitization to isoproterenol but not terbutaline. In marked contrast, neonatal hepatic cells demonstrated homologous desensitization to either single doses of or prolonged exposure to isoproterenol and terbutaline. In fact, there was increased sensitivity to agents that increase adenyl cyclase through nonadrenergic mechanisms. These results suggest that the resistance of neonatal myocytes to desensitization is the result of processes downstream from the adrenergic receptor and may involve developmental changes in the expression of adenyl cyclase isoforms or the compartmentalization of PKA activity.
Another area of difference is the handling of intracellular calcium. In the adult heart, the majority of released calcium is derived from the sarcoplasmic reticulum, but this is less so in the neonatal heart.82 Calcium flux across the sarcolemma is the predominant source of calcium utilized in excitation-contraction coupling. Neonatal rabbit hearts express more NCX protein than do adult hearts. With maturation, expression of NCX decreases, accompanied by enhanced contractility. In addition, differences between neonatal and adult hearts could be demonstrated on the concentration-response curves for ouabain (dP/dt min) and calcium (dP/dt min), but not for isoproterenol.94 An experimental agent that blocks the NCX did not increase contractility in the neonatal heart, although it did have an inotropic effect on adult myocardium.94 The role of voltage-gated calcium channels as the source of intracellular calcium in the neonatal myocardium is unclear. Studies suggest that these channels are the major source of intracellular calcium, although their exact role is unclear.82,99,100
Developmental differences also may exist in the expression of phosphodiesterase (PDE) isoforms. In a model of rabbit ventricular myocytes, administration of IBMX (a nonselective PDE inhibitor) and rolipram (a PDE type IV inhibitor) increased intracellular calcium currents in neonatal cells both at baseline and in response to isoproterenol but not in adult cells. In contrast, milrinone (a PDE type III inhibitor) increased intracellular calcium currents at baseline and in response to isoproterenol only in the adult cells.101 Thus it would appear that in the neonatal myocardium, PDE type IV may be the dominant isoenzyme that regulates intracellular calcium currents.
Developmental changes also may involve the peripheral vascular system. In developing swine, Gootman102 found that the peripheral vascular response to several adrenergic agonists developed at different rates and that complete reflex integration was not present at birth. He predicted that overall responses to stress or to treatment with these agents would be age dependent. Gootman’s physiological studies are complemented by receptor-binding studies that indicate that developmental changes exist in the adrenergic receptor content of a variety of organs, although more recent work indicates that lymphocyte β-adrenergic receptors are fully mature and functional at birth.103,104 Structural and ultrastructural differences also exist between immature and mature hearts. These differences include reduced ventricular compliance, greater ventricular interdependence, and a reduction in the ratio of myocardial contractile to noncontractile protein in the immature heart. The net effect is that the immature myocardium neither responds to nor tolerates volume loading as well as the adult heart. In addition to this diminished “preload reserve,” the baseline heart rate of infants and children is quite high, which limits the extent to which tachycardia can augment cardiac output before diastolic filling is compromised.
The combination of impaired preload reserve, limited chronotropic reserve, and reduced sensitivity of the heart and peripheral vasculature to adrenergic agents implies that the response of the immature organism to the infusion of inotropic and vasopressor agents may differ from the pattern noted in adults.105,106
Sympathomimetic Amines
Virtually all sympathomimetics currently used to treat hemodynamic problems are catecholamines. This class includes the endogenous compounds epinephrine, norepinephrine, and dopamine and the synthetic products isoproterenol and dobutamine. Catecholamines have a β-phenylethylamine core with hydroxyl (OH) substituents at the 3 and 4 aromatic ring positions (Figure 25-8). Minor differences in molecular substitution about the N-terminus or the α or β carbons produce marked differences in activity. Structure-activity relationships are complex for the catecholamines and have been reviewed; it is possible to generalize by noting that increasing size of the substituent on the amino group enhances β-adrenergic activity, whereas decreasing size is associated with α-adrenergic selectivity.107 Tyrosine serves as the base compound for the synthesis of catecholamines. Tyrosine hydroxylase catalyzes the conversion of tyrosine to dopa, which undergoes decarboxylation, producing dopamine. Dopamine β-hydroxylase converts dopamine to norepinephrine. In the adrenal medulla, norepinephrine is converted to epinephrine by n-methyl transferase (Figure 25-9).
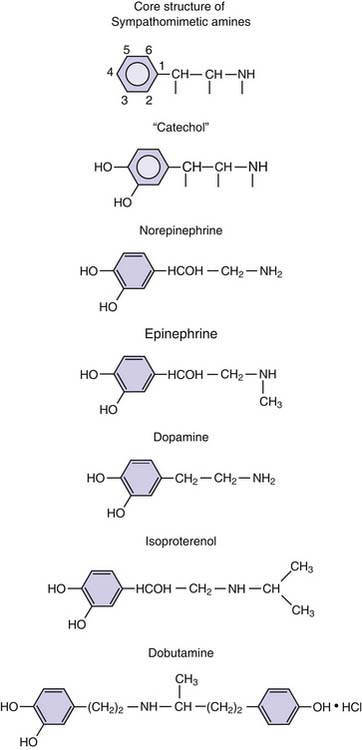
Figure 25–8 Chemical structure of the catecholamines.
(Modified from Chernow B, Rainey T, Lake R: Endogenous and exgoenous catecholamines in critical care medicine, Crit Care Med 10:409, 1982.)
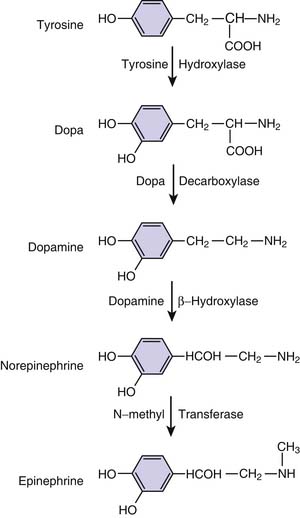
Figure 25–9 Biosynthetic pathways of the endogenous catecholamines.
(Modified from Chernow B, Rainey T, Lake R: Endogenous and exgoenous catecholamines in critical care medicine, Crit Care Med 10:409, 1982.)
Catecholamines are subject to several different elimination processes.107a Infused dopamine provides an example in which elimination occurs through a variety of processes. A small proportion is excreted unchanged in the urine. It is likely that a proportion undergoes neuronal reuptake. The principal means of elimination appears to be O-methylation by catechol O-methyltransferase (COMT) to form metanephrines, followed by either sulfoconjugation (by phenolsulfotransferase) or by deamination (by monoamine oxidase [MAO]) to homovanillic acid.108 Substitution at the α carbon determines the rate of deamination by MAO.109 The contribution of these pathways to total body clearance of catecholamines varies with age and the particular circulatory bed. In newborn lambs, the lungs accounted for 35% of the total body clearance of norepinephrine and 15% of the clearance of epinephrine. Inhibition of MAO by desipramine decreased pulmonary clearance to near zero and decreased total body clearance of norepinephrine and epinephrine clearance by 51% and 30%, respectively.110 In adult rabbits, inhibition of COMT and MAO simultaneously decreased pulmonary clearance of norepinephrine, epinephrine, and dopamine but had only minor effects on total body clearance.111 Inhibition of COMT did not change extracellular levels of catecholamines in the CNS.112 Furthermore, individual differences in COMT activity are not well correlated with dopamine clearance.113 The liver and gut have been shown to clear between 30% to 52% of the circulating norepinephrine and epinephrine.114,115 It is likely that processes or drugs that disturb these routes of elimination will decrease the overall metabolic clearance of catecholamines. Organ dysfunction associated with critical illness is known to increase the blood concentration of dopamine during a given infusion rate of the compound. For example, with liver dysfunction the clearance of dopamine is reduced.116
Dopamine
Basic Pharmacology
In the enzymatic pathway leading from tyrosine to epinephrine (Figure 25-9), decarboxylation transforms L-dopa to dopamine. Dopamine is a central neurotransmitter and is also found in sympathetic nerve terminals and in the adrenal medulla, where it is the immediate precursor of norepinephrine. In healthy persons, plasma levels of dopamine are in the range of 50 to 100 pg/mL.
Clinical Pharmacology
Dopamine simulates dopamine (D1 and D2) receptors located in the brain and in vascular beds in the kidney, mesentery, and coronary arteries (Table 25-3).117 It also stimulates α and β receptors, although the compound’s affinity for these receptors is lower. D1 receptors are coupled to Gs and thus enhance adenylate cyclase and produce a rise in cAMP, which evokes vasodilatation. This increases blood flow to these organs and may enhance renal solute and water excretion by the kidney. Dopamine also modulates release of aldosterone and prolactin (D2 receptors), which also may affect renal solute clearance.118 The physiological role of dopamine has been extensively reviewed.119,120
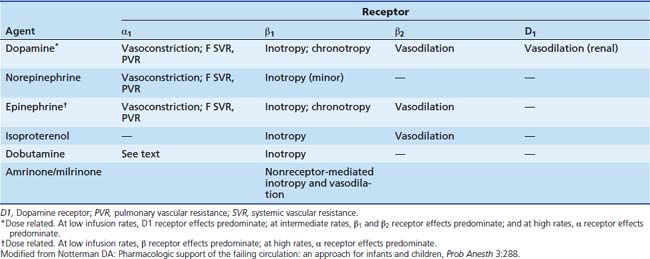
In healthy adult volunteers, infusion rates between 1 and 10 μg/kg/min increase stroke volume and cardiac output without major effect on heart rate or blood pressure.121–123 These infusion rates are associated with plasma concentrations of 50 to 100 ng/mL. Low infusion rates augment renal sodium excretion; intermediate rates (10 μg/kg/min) produce chronotropic and inotropic effects, and still higher infusion rates increase vascular resistance.124 Renal blood flow, glomerular filtration rate, and sodium excretion are maintained or even increase during dopamine infusion in patients with poor cardiac output. Work by Gundert-Remy and colleagues125 in healthy adults indicated that even at a relatively low infusion rate of 400 μg/min (5 to 6 μg/kg/min) there is significant augmentation of heart rate and blood pressure that likely plays a role in improved renal function.
Evidence for the view that infants display reduced sensitivity to dopamine is not conclusive. In support of reduced sensitivity, Perez and associates126 found that in critically ill neonates, infusion rates of 50 μg/kg/min did not cause clinically evident impairment of cutaneous or renal perfusion. Some experimental evidence for diminished sensitivity to dopamine in infants also exists; however, this evidence is limited to studies in immature animals. A contrary observation was made by Padbury and co-workers,127 who measured cardiac output in a group of infants and found that mean blood pressure increased at doses of 0.5 to 1 μg/kg/min, whereas heart rate increased beyond 2 to 3 μg/kg/min. Cardiac output and stroke volume increased before heart rate, and systemic vascular resistance (SVR) did not change within the range of dopamine infusion rates (0.5 to 8 μg/kg/min). The threshold values obtained were 14 ± 3.5 ng/mL for increase in mean blood pressure, 18 ± 4.5 ng/mL for increase in systolic blood pressure, and 35 ± 5 ng/mL for increase in heart rate. The steady-state concentration at infusion rates between 1 and 2 μg/kg/min was 16.5 ± 3.4 ng/mL. Thus newborns may exhibit clinical response at doses as low as 0.5 to 1 μg/kg/min. Seri and colleagues128 demonstrated an increase in blood pressure without a change in heart rate in premature infants. Doses ranged from 2.5 to 7.5 μg/kg/min.128 Interestingly, in this study, a greater increase in blood pressure was associated with a lower gestational age. The authors attributed this finding to the enhanced α adrenergic sensitivity of the immature myocardium.
Infused dopamine crosses the blood-brain barrier in preterm neonates but was shown not to increase blood flow velocity in the middle cerebral artery; dopamine inhibits the release of prolactin, thyrotropin, growth hormone, and the gonadotropins, but the clinical significance of these effects is unclear.128–130 A study in 35 very low birth weight neonates with hypotension randomly assigned to receive dopamine or dobutamine confirmed an inhibitory dopamine effect on prolactin, thyroid-stimulating hormone, and thyroxine secretion. However, hormone levels rapidly normalized following the discontinuation of the dopamine infusion, and the authors speculated that there would be no long-term consequences.131 This study also confirmed that dopamine is significantly more effective for blood pressure support in this population than dobutamine. The effectiveness of dopamine for blood pressure support also was shown in a separate study in 17 premature neonates with hemodynamically significant patent ductus arteriosus. A mean dopamine infusion rate of 8 μg/kg/min significantly increased systemic arterial pressure (30 ± 3 to 41 ± 5 mm Hg; P < .05), pulmonary artery pressure (25 ± 5 to 32 ± 8 mm Hg; P < .05), and superior vena caval blood flow (130 ± 40 to 170 ± 44 mL/kg/min; P < .05), leading to a decrease in left-to-right ductal shunting of blood flow.132
Low infusion rates of dopamine are frequently employed to augment renal function during critical illness.133 Although evidence exists that this treatment may increase the fractional excretion of sodium and the creatinine clearance, a large randomized, double-blind placebo control study of low-dose dopamine (2 μg/kg/min) administered to critically ill patients at risk of renal failure did not show any benefits.134,135 The study group and the control group did not differ in peak creatinine concentration, need for renal replacement therapy, length of ICU or hospital stay, or mortality. Seri and colleagues128 demonstrated an increase in urine output among premature infants associated with an increase in blood pressure and decrease in renal vascular resistance with dopamine, although they did not take into account the role of postnatal physiologic changes in urine volume. Use of low-dose dopamine in pediatric and neonatal ICUs to augment renal function is not uncommon despite the lack of evidence to suggest a beneficial effect.136
Pharmacokinetics
Plasma dopamine clearance ranges from 60 to 80 mL/kg/min in normal adults and is lower in patients with renal or hepatic disease.116,137 In subjects with normal renal function, the elimination half-life of infused dopamine is approximately 2 minutes.138 Among critically ill children, the elimination half-life is 26 ± 14 minutes, and in neonates the elimination half-life is 5 to 11 minutes.139,140 Age has a striking effect on clearance of dopamine, and clearance in children younger than 2 years of age is approximately twice as rapid as it is in older children (82 vs. 46 mL/kg/min).116 Wide interindividual variations in the rate of dopamine clearance have been reported in critically ill children as well as in healthy adults.141,142 Dopamine clearance may also decrease after 24 hours of continuous infusion.7 Allen and colleagues113 confirmed that during the first 20 months of life, clearance of infused dopamine decreased by almost 50%, with an additional 50% decrease from ages 1 to 12 years. Another study did not show a correlation between age and dopamine clearance, although the patients in this study had a mean age of 37 months.141 Age-related differences in COMT activity cannot account for the higher dopamine clearance values exhibited in neonates.113 Banner and colleagues143 studied the pharmacokinetics of infused dopamine in 15 patients ranging in age from 3 days to 8 years and noted nonlinear behavior, possibly resulting from saturable plasma protein binding in neonates. Although dopamine clearance correlated significantly with body weight, the authors questioned the utility of evaluating total body clearance in this age group. Differences in the rate of sulfoconjugation as a route of elimination also may contribute to the wide variations in the clearance of dopamine in critically ill children.113,116,141 The possible role of concomitantly administered dobutamine on the clearance of dopamine has been suggested by some authors, but an in vitro study showed that although dopamine and dobutamine are competitors for both COMT and MAO, the concentrations achieved under clinical situations are unlikely to produce clinically significant levels of inhibition.140,144 A pharmacokinetic difference between children and infants, rather than a difference in receptors or myocardial sensitivity, may account for the observation that infants require and tolerate higher infusion rates. Dopamine crosses the human placenta, but the effect on the fetus is not known. The pharmacokinetics of dopamine and other cardiovascular drugs has been reviewed.145
Clinical Role
Clinicians use dopamine to enhance renal function and to exploit its inotropic and vasopressor properties. Dopamine has been shown to be an effective inotropic and vasopressor agent in neonates and infants with a variety of conditions associated with circulatory failure, including hyaline membrane disease, asphyxia, sepsis syndrome, and cyanotic congenital heart disease.146,147 Very few trials of pharmacological therapy for hypotension in the neonate have been conducted. Osborn and colleagues148 found that although dopamine increases blood pressure, dobutamine produced a greater increase in blood flow, as judged by superior vena cava flow. Four other trials showed that dopamine was more effective than dobutamine in treating hypotension, but there was no difference in mortality.149 Fewer data evaluating the efficacy of dopamine in older children are available. However, it remains a mainstay of pharmacologic support for the child with inadequate perfusion. Dopamine is recommended as the first-line agent for children with fluid refractory septic shock and has been recommended for adults as well, but this topic is debated.150–152 Dopamine also may be appropriate for children with mild impairment of myocardial function and hypotension after resuscitation from cardiac arrest.153,154 Severe impairment of vascular tone or of cardiac contractility suggest the need for other agents. Children with primary myocardial disease not complicated by frank hypotension will benefit from a more selective inotropic agent such as dobutamine or milrinone. Infusion rates of dopamine needed to improve signs of severe myocardial dysfunction may be associated with troublesome tachycardia or dysrhythmia and may increase myocardial oxygen consumption disproportionately to myocardial perfusion.
Although dopamine is used extensively following cardiac surgery, reports indicate that dopamine is less effective following cardiac surgery in infants than it is in older children or adults.155 Lang and colleagues156 treated five children with dopamine following cardiac surgery. For the group as a whole, hemodynamic improvement did not occur at infusion rates less than 15 μg/kg/min. When cardiac output did increase, it was attributed to an increase in heart rate rather than to improved stroke volume. Another study indicated that following cardiac surgery, dopamine and dobutamine have similar inotropic efficacy, but that dopamine was associated with pulmonary vasoconstriction at dosages greater than 7 μg/kg/min in the absence of α-adrenergic blockade.157 Dopamine at 10 μg/kg/min improved right ventricle (RV) function after RV injury in a young swine model.158 Increased RV ejection fraction with a decrease in end-systolic RV volume was noted in premature hypotensive infants treated with dopamine.159 To treat shock associated with hypotension, therapy is initiated with an infusion rate of 5 to 10 μg/kg/min. The rate of infusion is increased in steps of 2 to 5 μg/kg/min, guided by evidence of improved blood flow (skin temperature, capillary refill, sensorium, and urine output) and by restoration of a blood pressure that is appropriate for age. Infusion rates greater than 25 to 30 μg/kg/min of dopamine are not customary, even if they maintain a “normal” blood pressure. At infusion rates of this magnitude, the effect on blood pressure is likely to represent an increase in SVR (α-adrenergic activation) rather than cardiac output. A requirement for a dopamine infusion of this magnitude suggests that the physician reexamine the physiological diagnosis or select a different agent, such as epinephrine or norepinephrine.
Adverse Effects
Dopamine toxicity is mainly cardiovascular, resulting in tachycardia, hypertension, and dysrhythmia. Dopamine is less likely to produce severe tachycardia or dysrhythmias than either epinephrine or isoproterenol.160 With the possible exception of the bipyridines, all inotropes increase myocardial oxygen consumption because they increase myocardial work. If the resulting increase in oxygen consumption is balanced by improved coronary blood flow, the net effect on oxygen balance is beneficial. When shock is caused or complicated by myocardial disease, improved myocardial contractility may reduce preload and afterload, improve coronary perfusion pressure (increase oxygen supply), and prolong diastolic coronary perfusion by reducing heart rate. If the same drug is administered to a patient with normal myocardial contractility, the result may be an increase in cardiac oxygen consumption without an increase in oxygen delivery to the myocardium. Tachycardia, by both increasing oxygen consumption and shortening diastole, is a particular burden. Thus the effect of dopamine on myocardial oxygen balance is better than that of isoproterenol, but not as good as dobutamine, amrinone, and milrinone.161 Dopamine depresses the ventilatory response to hypoxemia and hypercarbia by as much as 60%.162 Dopamine (and other β-agonists) decrease PaO2 by interfering with hypoxic vasoconstriction.163 In one study dopamine increased intrapulmonary shunting in patients with acute respiratory distress syndrome (ARDS) from 27% to 40%.164 The effect of dopamine on perfusion to the splanchnic bed is widely debated. Evidence suggests that dopamine may increase splanchnic blood flow during sepsis and after cardiac surgery, increase gastric pH, and lead to less lactate production than epinephrine.165–168 However, dopamine may increase blood flow or oxygen delivery but reduce oxygen consumption in patients with sepsis.169,170 In infants and children who have undergone cardiac surgery, dopamine may have several endocrinologic effects. Prolactin levels are decreased, the pulsatility of growth hormone is decreased in infants, and thyrotropin levels are decreased.118 Dopamine can cause or worsen limb ischemia, gangrene of distal parts and entire extremities, and extensive loss of skin.171 Infusion rates as low as 1.5 μg/kg/min have been associated with limb loss. Because dopamine promotes release of norepinephrine from synaptic terminals (and is also converted to norepinephrine in vivo), it is more often associated with limb ischemia than other adrenergic compounds. Extravasations of dopamine should be treated immediately by local infiltration with a solution of phentolamine (Regitine, 5 to 10 mg in 10 mL of normal saline solution) administered with a fine hypodermic needle.172
Preparation and Administration
Dopamine hydrochloride is available in 5-mL vials at concentrations of 40 mg/mL, 80 mg/mL, and 160 mg/mL, and as 250 mL or 500 mL premixed solutions for infusion at concentrations of 0.8 mg/mL, 1.6 mg/mL, and 3.2 mg/mL in 5% dextrose.139 Dopamine is administered by central vein to avoid the risk of skin injury resulting from extravasation. In an emergency, dopamine may be administered through an intraosseous needle.173 Table 25-4 and Table 25-5 provide information regarding preparation of infusions and compatibility.174,175 Table 25-4 presents a dilution method sometimes referred to as the “rule of 6.” Evidence suggests that preparation of medicated infusions on patient care units, rather than in a hospital pharmacy, is associated with an increased incidence of medication errors. Therefore several groups and agencies now discourage use of the rule of 6 or any other system that involves nonstandard medication concentrations. Practitioners should consult the policies of their institution before prescribing any of the drugs listed in Table 25-4. Dopamine is not compatible with some of the 3-in-1 solutions utilized for parenteral nutrition or with sodium bicarbonate.176 Dopamine is stable in solutions of 5% dextrose or normal saline solution for 24 hours to 84 hours.176a,177
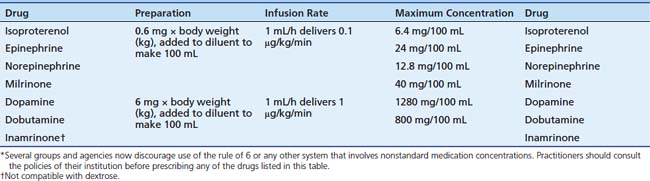
Table 25–5 Compatibility of Vasoactive Drugs with Commonly Used Continuous Infusions
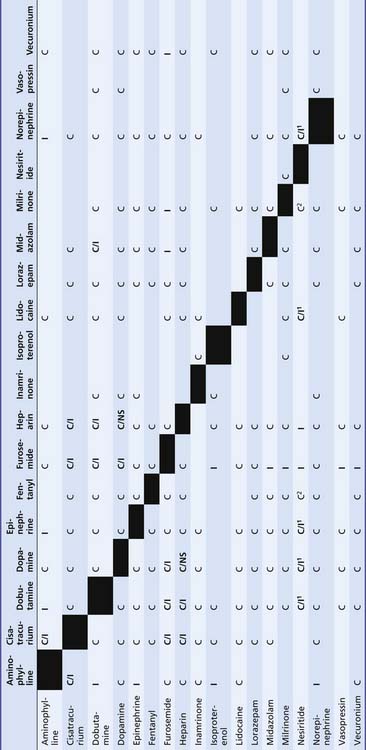 |
C, Compatible; C/I1, may be unstable at higher concentrations of additives; C/NS, this combination more stable in normal saline solution (NS) than in D5W because of an exothermic reaction; I, incompatible.
∗ Norepinephrine = More stable in D5W at higher concentrations because of its high level of acidity (pH 3).
1, Incompatible with products containing bisulfite antioxidants.
2, Stable for 4 hours.
From Trissel LA: Handbook on injectable drugs, ed 15, Bethesda, MD, 2009, American Society of Health-System Pharmacists.
Interactions
Dopamine is metabolized by MAO, and concurrent use of a MAO inhibitor (e.g., pargyline) potentiates its effect.107 In this rare circumstance the initial dosage of dopamine should be reduced to one tenth the usual dosage.178 Both α-adrenergic blockers and β-adrenergic blockers antagonize the effects of dopamine; dopamine antagonists such as metoclopramide or haloperidol also may attenuate response to dopamine. An increase in dopamine infusion rate will overcome the receptor blockade if necessary.
Summary
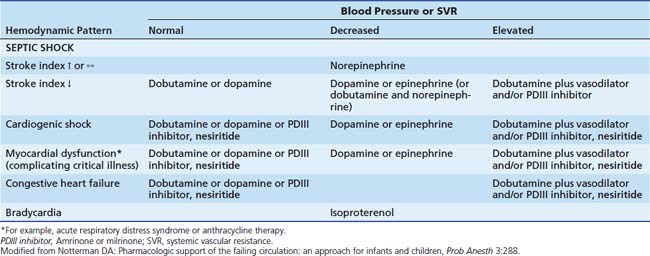
Dopamine is used to treat mild to moderate cardiogenic or distributive (septic; hypoxic-ischemic) shock associated with moderate degrees of hypotension. In the absence of hypotension, acute severe cardiac failure is treated with dobutamine or amrinone. When septic or cardiogenic shock is complicated by severe hypotension, epinephrine, or norepinephrine is preferred, depending on hemodynamic measurements (Table 25-6).
Norepinephrine
Basic Pharmacology
Dopamine is hydroxylated at the β-carbon to produce norepinephrine, the principal neurotransmitter of the sympathetic nervous system (Figure 25-8 and Figure 25-9). Because there is no substituent on the N (amino)-terminus, norepinephrine has little β2 activity and is considerably less potent at that receptor than epinephrine.107 It is a moderately potent α and β1 agonist.
Clinical Pharmacology
Infusion in normal subjects elevates SVR because α-adrenergic stimulation is not opposed by β2 stimulation.107 Reflex vagal activity reduces the rate of sinus node discharge, thereby blunting the expected β1 chronotropic effect. In normal subjects renal, splanchnic, and hepatic blood flows decrease. The increase in afterload may augment coronary blood flow. This effect may be enhanced by α-adrenergic receptors located in the coronary arteries, although in coronary arteries from explanted human hearts, the vasodilatation in response to norepinephrine was mediated via β2 receptors.179 Norepinephrine does have inotropic effects on the heart, mediated via α1 and β1 receptors. The proportion of inotropic response related to α1 stimulation may be affected by the pressure load on the right ventricle.180 In the failing heart, the relative contribution from each type of adrenergic receptor appears to be equal.181 In the isolated rat heart, the inotropic effects of norepinephrine are diminished in the presence of a nitric oxide synthetase inhibitor but restored when a nitric oxide donor is present.182 Interestingly, the contractile effects of norepinephrine in the rat are lost when it is pretreated with peroxynitrite.183 In healthy volunteers norepinephrine produces a decrease in creatinine clearance because of the effect on renal blood flow; however, in patients with hypotension, the improvement in global perfusion may actually produce an increase in urine output.184
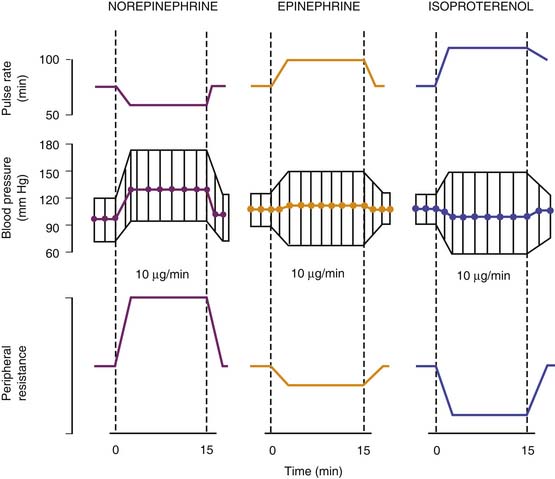
The acute hemodynamic effects of norepinephrine are compared with those of epinephrine and isoproterenol in Figure 25-10. Experience in critically ill children indicates that the hemodynamic responses are not different from those observed in adults. Use of norepinephrine in 18 neonates with persistent pulmonary hypertension–associated heart dysfunction revealed significant increases in systemic blood pressure (33 ± 4 to 49 ± 4 mm Hg; P < .05) and left ventricular output (172 ± 79 to 209 ± 90 mL/kg/min; P < .05), along with improvement in postductal transcutaneous oxygen saturation (89% ± 1% to 95% ± 4%; P < .05), pulmonary-to-systemic blood pressure ratio (0.98 ± 0.1 to 0.87 ± 0.1; P < .05), and a 20% increase in the velocity of pulmonary artery blood flow (P < .05).185 In a separate study in 22 neonates with septic shock refractory to fluid support, dopamine, and/or dobutamine, norepinephrine (mean infusion rate 0.5 ± 0.4 μg/kg/min) significantly increased mean arterial blood pressure (36 ± 5 to 51 ± 7 mm Hg; P < .001) and urine output (1 ± 0.5 to 1.7 ± 0.4 mL/kg/hr; P < .05), while decreasing blood lactic acid concentrations (4.8 ± 2.3 to 3.3 ± 1.8 mmol/L; P < .01).186
Pharmacokinetics
Basal plasma levels of norepinephrine are much higher than basal plasma levels of epinephrine (250 to 500 vs. 20 to 60 pg/mL). The minimum concentration at which norepinephrine produces detectable hemodynamic activity is at least 1500 to 2000 pg/mL, suggesting that endogenous plasma norepinephrine simply represents “spillover” from sympathetic activity and that norepinephrine is not a true hormone.187 The clearance of norepinephrine in healthy adults is 24 to 40 mL/kg/min, with the half-life averaging 2 to 2.5 minutes.188 The clinical effect of norepinephrine ceases within 2 minutes of the infusion being stopped.188 Little pharmacokinetic information in children is available. Norepinephrine is inactivated by reuptake into nerve terminals, with some elimination occurring by enzymatic degradation in the liver, adrenal glands, and kidney, either by methylation to normetanephrine (by COMT) or by oxidative deamination.189 Most of the metabolites formed by either MAO or COMT are then reduced or oxidized further. 3-Methoxy-4-hydroxymandelic acid is the major metabolite in the urine.107
Clinical Role
Norepinephrine improves perfusion in children with low blood pressure and a normal or elevated cardiac index. Septic shock is the usual context in which norepinephrine is beneficial. Norepinephrine is administered only after intravascular volume repletion and is best guided by estimates of cardiac output and SVR. Very little experience on the use of norepinephrine to treat distributive shock in children has been published; however, publications concerning adult patients provide a rationale for using this agent in patients with hypotension that is unresponsive to volume repletion and infusion of dopamine.190–193a In children, norepinephrine is the recommended agent for warm shock refractory to fluid loading and dopamine.150 A prospective, unblinded randomized study (in adults) indicates that norepinephrine is superior to dopamine for treating hypotension and other hemodynamic abnormalities associated with hyperdynamic septic shock. In this study, the average infusion rate for norepinephrine was 2.7 μg/kg/min compared with the average dopamine dose of 22 μg/kg/min.194 Others have reported that somewhat lower average dosages (0.4 μg/kg/min) were effective in adults with sepsis.195 Coronary and renal blood flow increased in lambs at a dose of 0.4 μg/kg/min while mesenteric blood flow decreased.196 Thus titration is important and may entail fairly rapid escalation of dosage. Norepinephrine produces increases in SVR, arterial blood pressure, and urine flow. It is most valuable in the context of tachycardia because infusion of the drug does not produce significant elevation of heart rate and may even lower heart rate through reflex mechanisms. In a study of adults with abdominal sepsis, norepinephrine infusion was associated with increases in systemic blood pressure and SVR. Stroke volume increased as heart rate declined. Cardiac index did not change, although creatinine clearance increased substantially.195 Norepinephrine has been shown to improve right ventricular performance in adults with hyperdynamic septic shock.194
The usual starting dosage is an infusion of 0.1 μg/kg/min (Table 25-7), with a goal of elevating perfusion pressure so that the flow to vital organs is above the threshold needed to meet metabolic requirements.197 Arbitrary values of SVR or blood pressure are not appropriate end points for therapy, and the lowest infusion rate that improves perfusion as judged by skin color and temperature, mental status, urine flow, and reduction in plasma lactate level should be used.198
Table 25–7 Suggested Infusion Rates for Inotropic and Vasopressor Agents (μg/kg/min)
| Agent | Clinical Indication | |
|---|---|---|
| Inotropic | Pressor | |
| Dopamine | 2-15 | >12 |
| Epinephrine | 0.05-0.5 | 0.10-1 |
| Norepinephrine | 0.05-1 | |
| Vasopressin | 0.0002-0.002∗ | |
| Dobutamine | 2.5-20 | |
| Inamrinone† | 5-10 | |
| Milrinone† | 0.25-0.75 | |
| Nesiritide† | 0.01 | |
| Isoproterenol | 0.05-1 | |
∗ mU/kg/min; the optimal dosage and infusion rate have not been established in children.
Other causes of distributive shock (e.g., vasodilator ingestion and intoxication with CNS depressants) also should respond to norepinephrine infusion when the predominant hemodynamic problem is low SVR and blood pressure.
Adverse Effects
The increase in afterload that norepinephrine produces should increase myocardial oxygen consumption, but norepinephrine may reflexively decrease heart rate, which should reduce oxygen consumption and improve diastolic coronary perfusion.107 Injudicious use of norepinephrine will lead to compromised organ blood flow. Norepinephrine infusion may elevate blood pressure yet not improve clinical indices of perfusion. This type of poor clinical response is usually associated with a low cardiac index, stroke volume, LV stroke work index, and an elevated pulmonary capillary wedge pressure.190,191 Employing excessive dosages or using norepinephrine to elevate blood pressure without improving perfusion may produce multiple organ system failure.






Full access? Get Clinical Tree


