Pharmacology of Perineural Analgesia
Brian A. Williams
Nigam Sheth
Sylvia Wilson
Lavinia Kolarczyk
Background and indications
Koller is credited with the use of cocaine for topical ophthalmic anesthesia (1884),1 Bier is credited for the use of cocaine for spinal anesthesia (1898),1 and Braun is credited for the use of epinephrine as an adjuvant to procaine (1900).2 In the past 20 years, novel perineural adjuvants (e.g., clonidine, buprenorphine, dexamethasone), have been combined with local anesthetics. These appear to be safe in preclinical laboratory models when combined with local anesthetics. The exception is midazolam which significantly worsens in vitro neurotoxicity. The local anesthetics discussed throughout this text book (bupivacaine, ropivacaine, levo-bupivacaine, mepivacaine, lidocaine, and chloroprocaine) can be more extensively reviewed in other current textbooks.
Physiology and pharmacology
Local anesthetics (LAs) are weak bases (nonionized). Only the nonionized isoform diffuses across the lipid nerve sheath of the peripheral nerve. The onset of action depends on the amount of drug in the nonionized form. Because of this, sodium bicarbonate (NaHCO3) speeds block onset by increasing LA pH and local tissue pH closer to the pKa of the LA. Conversely, acidosis or infection delays block onset by decreasing the pH both of LA and of local tissue. The ionized isoform acts intracellularly to block voltage-gated sodium (NaV) channels, thereby inhibiting impulse conduction and slowing depolarization. The transmembrane electrical threshold potential is not reached, and therefore the action potential is not propagated (Fig. 2.1).
In perineural use, LAs bind NaV channels in the axolemma, preventing NaV channels from opening and thus preventing action potentials. Different NaV channel types have differing affinities for LAs, which becomes imporTant when considering local anesthetic systemic toxicity. Specifically, cardiac NaV channel subtype 1.5 has remarkable affinity for bupivacaine, and this accounts for the potential for cardiac arrest with unwanted intravascular injection or absorption of bupivacaine. Peripheral nerves (NaV 1.7, 1.8, 1.9), dorsal root ganglia (NaV 1.7), and skeletal muscle (NaV 1.4) do not have significant, if any, presence of the NaV 1.5 channel.
It should be clearly understood that the conduction block mechanism of LA (at NaV channels) is not likely the only analgesic mechanism associated with LA. Likewise, LA activity at NaV channels does not contribute to the neurotoxic (i.e., nerve damage) risks of LA. Nerve damage risks can occur at generally accepted clinical doses via mechanisms unrelated to NaV channel activity.3 Axonal damage from LA can occur due to a variety of mechanisms including: (1) intrafascicular injection (which is theoretically
preventable with ultrasound-guided regional anesthesia by a well-trained practitioner), (2) with LA in high concentration, or (3) in situations of prolonged exposure (such as with a perineural catheter). More relevant is the potential for disruption of numerous cellular functions; primarily, intracellular calcium levels may play a central role.4 Specifically, there is disruption of cytoplasmic calcium signaling, leading to release of calcium from intracellular stores and leakage of calcium into the cell across the cell membrane. Ultimately, excess cytoplasmic calcium leads to neuronal death from the activation of intracellular kinases and the disruption of electron transport in the mitochondria. Apoptosis (programmed cell death) is closely linked to these intracellular calcium alterations.3 It is critical to acknowledge that there is no evidence that ultrasound-guided regional anesthesia protects patients from potentially toxic alterations of intracellular calcium after peripheral nerve block.
preventable with ultrasound-guided regional anesthesia by a well-trained practitioner), (2) with LA in high concentration, or (3) in situations of prolonged exposure (such as with a perineural catheter). More relevant is the potential for disruption of numerous cellular functions; primarily, intracellular calcium levels may play a central role.4 Specifically, there is disruption of cytoplasmic calcium signaling, leading to release of calcium from intracellular stores and leakage of calcium into the cell across the cell membrane. Ultimately, excess cytoplasmic calcium leads to neuronal death from the activation of intracellular kinases and the disruption of electron transport in the mitochondria. Apoptosis (programmed cell death) is closely linked to these intracellular calcium alterations.3 It is critical to acknowledge that there is no evidence that ultrasound-guided regional anesthesia protects patients from potentially toxic alterations of intracellular calcium after peripheral nerve block.
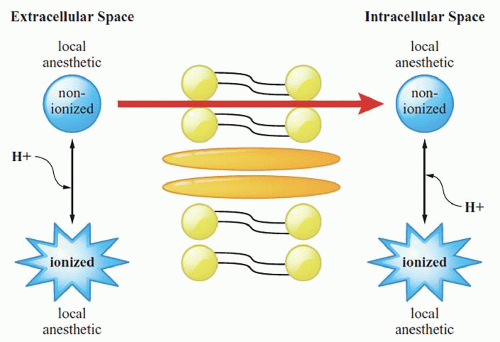 Figure 2.1. Any injected ionized isoform of the local anesthetic is rendered nonionized by tissue buffers and/or addition of NaHCO3. The nonionized molecule traverses the lipid bilayer membrane. Molecules are then reprotonated based on equilibration consTants, and the LA molecule ultimately binds into NaV channels when these channels are in an open or inactivated state (there is less affinity of these channels for LA molecules when the channels are in a resting state). |
The mechanisms of action for clonidine, buprenorphine, dexamethasone, and midazolam have not been as well described. Research is needed to determine the extent to which perineural adjuvants can reduce the overall dose-volume-concentration of the LA. Reducing total LA dose should be a primary objective in our subspecialty’s clinical and research agenda due to the known neurotoxicity of LA.
Clonidine-mediated perineural analgesia does not occur via agonist activity at the level of the alpha-2 adrenoreceptor; rather, it is mediated via the hyperpolarization-activated cation current.5 The same appears to be true for dexmedetomidine (based on in vivo preclinical models).6,7 It should be noted that the alpha-2 adrenoreceptor does mediate inflammatory responses of various white blood cells and appears to decrease perineural inflammation at the injection site of local anesthetic when alpha-2 agonists are co-administered with LA.8
Buprenorphine is presumably active after transaxonal influx at axon-level opioid receptors and adjacent axoplasmic G-protein coupling mechanisms, typical of opioid receptors, although this is not verified.
A corticosteroid (methylprednisolone) has been described to suppress the transmission in thin unmyelinated C-fibers but not in myelinated A-beta fibers.9 Whether this same mechanism, or other mechanisms, holds true for perineural dexamethasone remains to be determined.
Midazolam is profoundly cytotoxic when combined with ropivacaine and exposed to primary sensory neurons (rat) in culture.10 Perineural analgesia from bupivacaine enhanced by midazolam has not been mechanistically determined; possibilities include peripheral benzodiazepine receptors or peripheral GABAA receptors. The authors of this textbook chapter do not express or imply the endorsement of a combination of midazolam and LA given as a peripheral nerve block to patients. Restated, the authors do not recommend any patient study or off-label treatment strategy that involves the use of combined LA and midazolam for peripheral nerve blocks. In vitro combinations of clonidine, buprenorphine, dexamethasone, and midazolam are not neurotoxic in cell culture of primary sensory neurons, but combining midazolam and ropivacaine is profoundly and synergistically neurotoxic.10
Local Anesthetic and Adjuvant Technique
Drugs are freshly prepared, and all drugs should be preservative free.
Readers are referred to the recommended local anesthetic doses for each block described in this textbook. Generally, the adjuvant technique described in this chapter synthesizes clinical experience, clinical research to date, and most relevant, recent in vitro research addressing the relative neurotoxicity of LA versus adjuvants. Attention will be directed toward the strategic reduction of LA concentrations when perineural adjuvants are used. The multiple adjuvants described in the following text are based on the in vitro findings that clonidine-buprenorphine did not increase LA neurotoxicity more than did the use of LA alone.10 Also considered is the in vitro dose-response evidence of dexamethasone worsening LA neurotoxicity (Williams et al., manuscript in review, 2010-2011, described further in the section “Summary of Evidence”).
The use of clonidine, buprenorphine, or both as perineural analgesic adjuvants is unequivocally considered off-label, and all associated precautions in patient education and documentation are suggested. Clonidine is added in doses as low as 0.25 µg/kg or as high as 1.5 µg/kg. Patients assuming the sitting position for surgery (e.g., after interscalene block; shoulder surgery in beach chair position) should be dosed at the lowest dose range in this spectrum. Doses of 1 µg/kg clonidine or greater can lead to more likely potential concerns of hypotension, bradycardia, and sedation (especially in surgical positioning other than supine).
Buprenorphine is added in doses from 1.5 to 3.0 µg/kg. Doses of 2.5 to 3.0 µg/kg are typically reserved for opioid-tolerant patients, and buprenorphine used perineurally in these doses does not appear to create opioid withdrawal symptoms or reverse systemic opioid analgesia. Doses of 1.5 µg/kg are typically used in patients receiving interscalene nerve blocks; otherwise, 2 µg/kg is a dose that can typically avoid opioid-induced side effects (nausea, vomiting, pruritus, etc.). Multimodal antiemetic prophylaxis,11 which has been recommended for both general and regional anesthesia,12,13 is especially useful when perineural buprenorphine use is planned (e.g., oral perphenazine, 8 mg, and intravenous ondansetrondexamethasone, 4 mg each).
In the absence of perineural dexamethasone dose studies addressing doses less than 8 mg, these authors recommend that if the decision is made to study dexamethasone or to use dexamethasone off-label in routine clinical care, the dexamethasone dose per nerve does not
exceed 1 to 2 mg until dose-response neurotoxicity is further elucidated. Dexamethasone in 1 to 2 mg perineural doses extrapolated to cell culture (and combined with ropivacaine 2.5 mg/mL, clonidine 1 µg/mL, and buprenorphine 3 µg/mL) is likely neither cytoprotective nor cytotoxic when compared with plain ropivacaine.
exceed 1 to 2 mg until dose-response neurotoxicity is further elucidated. Dexamethasone in 1 to 2 mg perineural doses extrapolated to cell culture (and combined with ropivacaine 2.5 mg/mL, clonidine 1 µg/mL, and buprenorphine 3 µg/mL) is likely neither cytoprotective nor cytotoxic when compared with plain ropivacaine.
The use of combined LA and midazolam for peripheral nerve blocks is absolutely not recommended by these authors.
Summary of evidence
For this textbook chapter, the authors consider clonidine and buprenorphine to be textbook drugs,14,



Related posts:






Full access? Get Clinical Tree
