Chapter 7 Pharmacology of Pediatric Anesthesia
Developmental pharmacology
Developmental changes profoundly affect the clinical response to medicines. Dr. Abraham Jacobi, a founder of American pediatrics, recognized more than a century ago that children are not “miniature men and women, with reduced doses and the same class of disease in smaller bodies,” that pediatrics “has its own independent range and horizon,” and that age-appropriate pharmacotherapy was important (Kearns et al., 2003). More recently, as the immaturity of renal and metabolic systems has been recognized, the pharmacologic uniqueness of babies and infants has been specifically recognized (Anderson and Holford, 2008). Physical growth, development, organ maturation, physiologic changes, and coexisting disease that occur throughout the spectrum of development—from preterm newborn to adolescence and adulthood—profoundly influence drug pharmacokinetics and pharmacodynamics, and ultimately the panoply of both desirable and undesirable clinical responses. This chapter presents the basic pharmacologic principles relevant to understanding basic pediatric pharmacology in general, and that of pediatric anesthetic pharmacology in particular.
Medications that are commonly used in children are not regularly tested in children, and drug labeling often consists exclusively of adult data. Of the 140 new molecular entities of potential use in pediatrics, only 38% were labeled for use in children when they were initially approved (Tod et al., 2008). Much use of drugs in pediatrics, particularly in newborns and infants, is off-label use. About one third of drugs prescribed in office-based pediatric practice, two thirds of those prescribed in hospitals, and 90% of drugs used in pediatric intensive care units are used for indications other than those for which they have been approved (Tod et al., 2008). When data were submitted to the U.S. Food and Drug Administration (FDA) to support labeling changes intended to guide pediatric drug use, on average, only 2.2 pediatric studies were represented (Abdel-Rahman et al., 2007). A considerable amount of pediatric drug use is based on “extrapolation” (or worse) from adult dosing and use guidelines. A major address of pediatric pharmacology research in the past decade has focused on the challenge of characterizing developmental changes in pharmacokinetics and pharmacodynamics, as well as determining proper pediatric dosing guidelines, particularly the downward scaling of adult doses to children.
Physiology and Development
Growth and maturation are seminal features of pediatric development, and it is important to understand that they vary independently (Anderson and Holford, 2008; Rhodin et al., 2009; Sumpter and Anderson, 2009). Growth is an increase in size, often characterized by changes in weight. Maturation is a time-dependent phenomenon, often characterized by age. Traditionally, the common metric of maturation was postnatal age (PNA), the time since birth. With the superiority of neonatal intensive care and the survival of neonates as small as 500 g and 24 weeks’ gestation, who have clearly immature and widely variant degrees of development at birth, it is now well established that PNA is an inadequate metric of organ maturation, which actually begins before birth. Postconceptual age (PCA), the sum of gestational age (the period between conception and birth) and PNA, is a superior metric. Because of the inexactitude in determining the date of conception, the alternate metric of postmenstrual age (PMA) is often used instead. Particularly for infants and very small children, use of PCA or PMA rather than PNA is important in pediatric pharmacology and therapeutics.
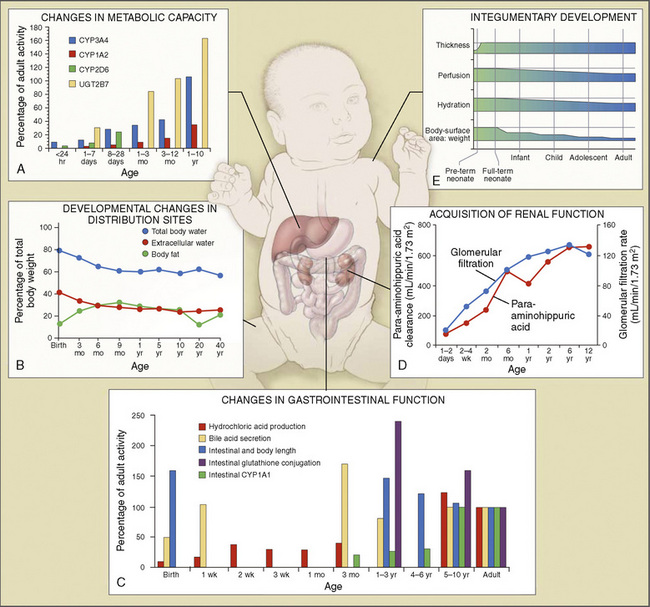
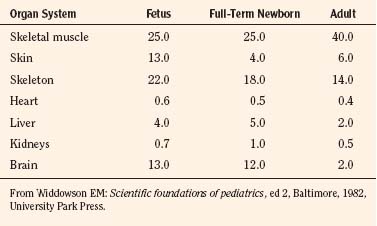
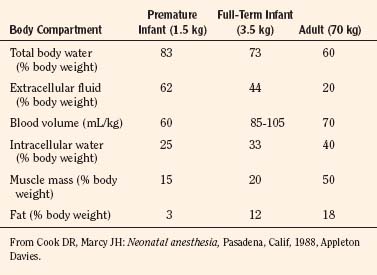
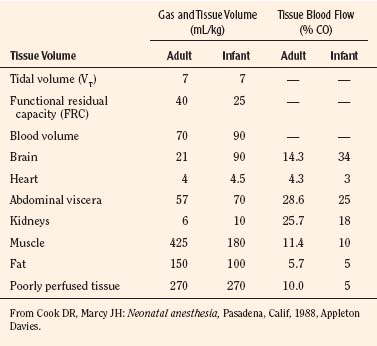
Body composition changes dramatically during growth and development (Anderson and Holford, 2008). Simply stated, compared with adults, infants have big heads, large torsos, and short, stumpy legs. Figure 7-1 depicts developmental changes in pediatric physiology that can affect drug disposition. Table 7-1 describes the changes in organ weight that occur during growth. The most significant changes with age are in total body water, the intracellular vs. extracellular distribution of body water, and muscle and fat mass (Tables 7-2 and 7-3). Total body water content constitutes 75% of body weight in the full-term newborn and 80% to 85% of body weight in the preterm neonate. This decreases to about 60% at 5 months and remains relatively constant until puberty. Extracellular water redistributes intracellularly during the first year of life. Extracellular fluid is 45% to 50% of body weight in premature and newborn infants, decreasing to 26% at 1 year and 18% in adults. In contrast, body fat increases with age, from 3% in premature neonates and 12% in full-term newborns, to 30% at 1 year of age. “Baby fat” is shed when toddlers start walking and drops to adult levels of about 18%, concomitant with an increase in muscle mass. A major consequence of body-composition changes may be differences in drug volume of distribution, with neonates and infants having larger volumes of water-soluble drugs, as described below.
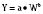
where Y is the biological characteristic, W is the body mass, and a and b are empirically derived constants. For physiologic functions such as cardiac output, metabolic rate, oxygen consumption, glomerular filtration rate (GFR), and pharmacologic functions such as drug clearance, the power exponent b is 0.75. For physiologic volumes such as blood volume, lung volume, tidal volume, and stroke volume, and pharmacologic volumes such as the volume of distribution, the power exponent is 1. For time-based physiologic functions such as circulation time, heart rate, and respiratory rate, and pharmacologic functions such as drug elimination half-life, the exponent is 0.25 (Johnson and Thomson, 2008). This allometric model may then be used to scale metabolic processes across size:
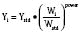
Pharmacokinetic Parameters
Volume of Distribution
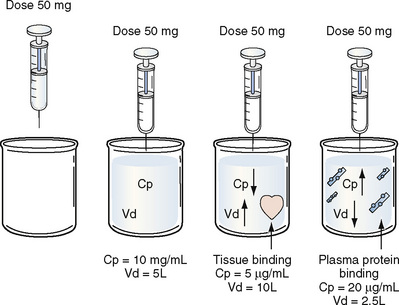
Volume of distribution (Vd) is a theoretic concept that relates the amount of drug in the body (dose) to the concentration (C) of drug that is measured (in blood, plasma, and unbound in tissue water). Volume of distribution is the volume of fluid “apparently” required to contain the total-body amount of drug homogeneously at a concentration equal to that in plasma (or blood) (Fig. 7-2):
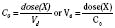
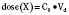
Clearance
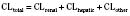
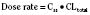
Renal Clearance
The kidney is the most important organ for elimination of water-soluble drugs and metabolites. This is particularly true for infants and small children, in whom hepatic metabolism is underdeveloped, and in whom age-dependent renal clearance is a major determinant of age-appropriate drug dosing (Kearns et al., 2003). Maturation of renal function, which occurs independently of PNA, is a dynamic process. Nephrogenesis begins between about weeks 5 and 9 of gestation and is complete by week 36, after which there are changes in renal blood flow (Kearns et al., 2003; Rhodin et al., 2009). GFR is approximately 5 mL/min in full-term neonates, but about one fifth that in preterm neonates. GFR increases with PMA, at a rate described as a maturation function (MF), based on the sigmoidal hyperbolic Hill equation, where PMA50γ is the maturation half-time, and γ is the Hill coefficient:
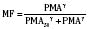
The rate of GFR maturation is nonlinear and maximum at about 48 weeks’ PMA. It varies with prematurity, with preterm neonates (33 to 34 weeks’ PMA) having a slower increase (14 mL/min per 1.73 m2 per week PMA) in the first few weeks of life than full-term neonates (39 to 41 weeks’ PMA; 94 mL/min per 1.73 m2 per week PMA). In general, healthy newborns’ GFR is about 30% of adult GFR and reaches adult values at about 8 to 12 months of age. Renal tubular function matures more slowly, reaching adult levels at about 12 to 18 months of age. For example, the dosing interval for tobramycin, which is eliminated renally, is 24 hours in full-term newborns but 36 to 48 hours in preterm newborns (Kearns et al., 2003).
Hepatic Clearance
For many drugs, including several used in anesthesia (e.g., sedative hypnotics, benzodiazepines, opioids, and neuromuscular blockers), hepatic clearance (CLH in the following formulas) is a major route of elimination (Wilkinson, 1987). Hepatic clearance is the product of liver blood flow (QH) and the hepatic extraction ratio (EH) of a drug.
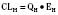
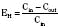
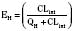
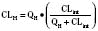
Since only an unbound drug is considered diffusible into liver cells (although in reality a simplification), EH also depends on the fraction of unbound drug in the blood (fu) (Baker and Parton, 2007). Therefore the three theoretic primary determinants of hepatic drug clearance are liver blood flow, intrinsic clearance (biotransformation), and plasma protein binding.
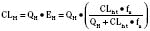
In reality, the important determinants of hepatic clearance are liver blood flow and the extraction ratio (Fig. 7-3). For drugs with a high extraction ratio (e.g., lidocaine, fentanyl, sufentanil, and propofol), hepatic clearance depends primarily on hepatic blood flow (because intrinsic clearance is so efficient, drug delivery to the liver becomes rate limiting). For low-extraction drugs (e.g., methadone, diazepam, and alfentanil), hepatic clearance is independent of hepatic blood flow and depends primarily on intrinsic clearance (metabolism).
Absorption
Although the predominant route of drug administration in pediatric anesthesiology is intravenous, the oral route is the most commonly used in children. Changes in physiologic processes that accompany normal growth and development can affect drug absorption (Kearns et al., 2003; Abdel-Rahman et al., 2007). In general, neonates and small children absorb drugs more slowly than older children and adults, resulting in delayed and lower peak drug concentrations. Neonatal gastric pH is relatively high (greater than 4) compared with older children and adults, thus acid-labile drugs (such as penicillin G) are more efficiently absorbed and have greater bioavailability. Conversely, weak acids (such as phenobarbital) may require larger doses because of reduced bioavailability. In the first few months of life, gastric emptying, intestinal motility, and intestinal drug transport increase.
Distribution
Plasma protein binding can exhibit developmental differences, although the clinical significance of protein binding in pharmacokinetics and pharmacodynamics remains unresolved (Benet and Hoener, 2002; Trainor, 2007). The primary binding proteins are albumin (for acidic drugs) and α1-acid glycoprotein (for basic drugs). Decreased plasma albumin and α1-acid glycoprotein concentrations in neonates may result in increased unbound (free) drug concentrations and hence pharmacologic effect.
Metabolism
Metabolism of a drug can result in bioactivation of an inactive prodrug, formation of an active or inactive metabolite, or occasionally a toxic metabolite. The liver is the primary site of drug metabolism, although the intestine can metabolize oral drugs before they reach the systemic circulation and extrahepatic metabolism (e.g., renal, blood, and other tissues) may be important for certain drugs (i.e., remifentanil and propofol). Phase I reactions (oxidation, reduction, and hydrolysis) chemically modify the drug structure to add, form, or uncover a functional group and render the molecule more water soluble. Phase II reactions (glucuronidation, sulphation, and glutathione conjugation) add an endogenous molecule to the drug or metabolite to render it even more water soluble for elimination. Typical pathways of drug metabolism are provided in Table 7-4.
TABLE 7-4 Pathways in Drug Metabolism
| Reaction | Examples |
| Phase I | |
| Oxidation reactions | Thiopental, methohexital |
| Aliphatic hydroxylation | Pentazocine, meperidine, glutethimide, doxapram, ketamine, chlorpromazine, fentanyl, propranolol |
| Aromatic | Lidocaine, bupivacaine, mepivacaine |
| Expoxidation | Phenytoin |
| O-Dealkylation | Pancuronium, vecuronium, codeine, phenacetin, methoxyflurane |
| N-Dealkylation | Morphine, meperidine, fentanyl, diazepam, amide local anesthetics, ketamine, codeine, atropine, methadone |
| N-Oxidation | Meperidine, normeperidine, morphine, tetracaine |
| S-Oxidation | Chlorpromazine |
| Oxidative deamination | Amphetamine, epinephrine |
| Desulfuration | Thiopental |
| Dehalogenation | Halogenated anesthetics |
| Dehydrogenation | Ethanol |
| Reduction Reactions | |
| Axo reduction | Fazadinium |
| Nitroreduction | Nitrazepam, dantrolene |
| Carbonyl reduction | Prednisolone |
| Alcohol dehydrogenation | Ethanol, chloral hydrate |
| Hydrolysis Reactions | |
| Ester hydrolysis | Ester local anesthetics, succinylcholine, acetylsalicyclic acid, propanidid amide local anesthetics |
| Phase II: Conjugation Reactions | |
| Glucuronamide | Oxazepam, lorazepam, morphine, nalorphine, codeine, fentanyl, naloxone |
| Sulfate | Acetaminophen, morphine, isoproterenol, cimetidine |
| Methylation | Norepinephrine |
| Acetylation | Procainamide |
| Amino acid | Salicyclic acid |
| Mercapturic acid | Sulfobromophthalein |
| Glutathione | Acetaminophen |
Modified from Tucker GT: Drug metabolism, Br J Anaesth 51:603, 1979.
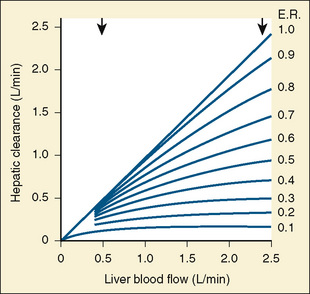
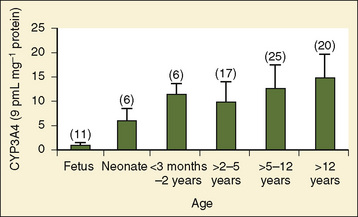
Comprehensive reviews of human drug metabolism are available, but a generalized overview is instructive for understanding developmental changes in biotransformation (Kramer and Testa, 2008; Pelkonen et al., 2008; Zanger et al., 2008). Cytochrome P450 (CYP) is the main oxidative (phase I) metabolizing enzyme system, and more than 50 human P450s have been identified, although only a small fraction are responsible for the majority of drug metabolism (Fig. 7-4) (Paine et al., 2006; Zanger et al., 2008). Individual CYPs are classified by their sequence evolution and amino-acid similarities (Ingelman-Sundberg et al., 2007; Zanger et al., 2008). Those with greater than 40% sequence homology are grouped in a family (designated by an Arabic number, e.g., CYP3); those with more than 55% homology are in a subfamily (designated by a letter, e.g., CYP3A), and individual CYPs are identified by a third number (e.g., CYP3A4). The majority of drugs in humans are metabolized by CYPs 1, 2, and 3 (particularly CYPs 1A2, 2B6, 2C8, 2C9, 2C19, 2D6 and 3A4, 3A5, and 3A7). CYPs can have numerous genetic variants, and there are several highly polymorphic CYPs. Allelic CYP variants are designated by an asterisk and number (e.g., CYP3A5*3, where the “wild-type” is always *1) (Ingelman-Sundberg et al., 2007; Zanger et al., 2008). CYPs can have varying degrees of substrate specificity, and some are very accommodating, like CYP3A, which metabolizes approximately one third to one half of all therapeutically used drugs.
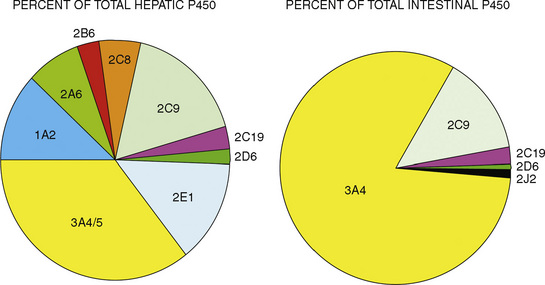
FIGURE 7-4 Cytochrome P450 isoform content in human liver and intestine.
(Data from Rowland-Yeo K et al.: Abundance of cytochromes P450 in human liver: a metaanalysis, Br J Clin Pharmacol 57:687, 2004; Paine et al.: The human intestinal cytochrome P450 “pie,” Drug Metab Dispos 34:880, 2006.)
The developmental pattern of biotransformation activity generally follows a hyperbolic curve with age that begins in fetal development, with hepatic drug metabolism and very low clearance before and during the first month. It reaches near adult levels at approximately 1 year, becomes maximum before puberty, and declines slightly into adulthood (Johnson and Thomson, 2008). There are, however, enzyme and isoform-specific patterns of developmental maturation of drug-metabolizing enzymes (Blake et al., 2005; Hines, 2008). Several pertinent examples are in the following paragraphs.
The CYP3A family is the most important drug-metabolizing enzyme. The important isoforms are CYP3A4 (the major CYP present in the adult liver and the intestine), CYP3A5 (which metabolizes very many of the CYP3A4 substrates with equal or diminished activity and is polymorphically expressed), and CYP3A7 (a fetal form that is usually less active than 3A4 or 3A5). CYP3A undergoes a “developmental switch” with CYP3A7, the predominant CYP3A expressed in fetal liver (Stevens, 2006; Hines, 2008). Indeed it is the most abundant of any CYP and is thought to be important in fetal steroid metabolism and homeostasis (Fig. 7-5, A). CYP3A7 expression decreases during gestation, declines further after birth, and is typically undetectable after the first year. CYP3A4 expression is low during development and then increases after the first 6 months of age. CYP3A metabolizes many drugs of importance in anesthesiology, including all of the fentanyl series opioids (except remifentanil), most benzodiazepines, and local anesthetics. Although high in abundance, CYP3A7 has low activity toward many drugs (1% to 3% of the activity of CYP3A4 towards midazolam and alfentanil) (Björkman, 2006). Therefore, CYP3A-catalyzed drug metabolism and clearance increases with age. For example, the hepatic extraction of midazolam is only 0.04 in neonates, compared with 0.38 in adults (Björkman, 2006). Midazolam clearance is similarly very low in neonates, particularly in preterm infants. The age-dependent maturation of midazolam clearance is shown in Figure 7-5, B.
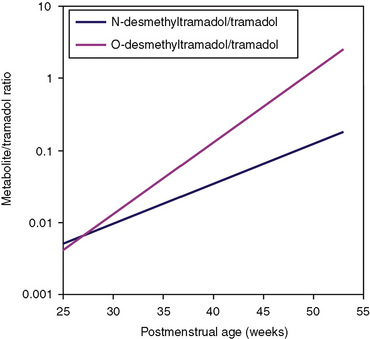
Other hepatic CYP isoforms also demonstrate maturation of expression. CYPs 1A2, 2C9, 2C19, 2D6, and 2E1 are expressed minimally or not at all in the fetus, but substantial increases occur after birth. CYP2D6 metabolizes about one fourth of all drugs, including antidepressants, antipsychotics, β-blockers, and numerous orally administered opioids, most notably catalyzing bioactivation of the inactive prodrugs codeine and tramadol (to the pharmacologically active metabolites morphine and O-desmethyltramadol, respectively) (Ingelman-Sundberg et al., 2007; Madadi and Koren, 2008). Isoform-dependent maturation of CYP enzymes in infants is exemplified by the development of tramadol metabolism and disposition, which shows that CYP2D6-catalyzed O-demethylation matures faster than CYP3A-catalyzed N-demethylation (Fig. 7-6). CYP1A2 metabolizes caffeine and theophylline, two methylxanthines that are commonly used in pediatrics. CYP1A2 is essentially absent in fetal liver and minimally active in neonates, such that 85% of caffeine is eliminated unchanged renally in neonates (Cazeneuve et al., 1994). CYP1A2 activity matures rapidly thereafter, reaching adult values by age 6 months, and caffeine clearance in these children reflects primarily hepatic demethylation (Kearns et al., 2003).
Compared with hepatic CYP enzymes, the ontogeny of CYPs in the intestine is far less understood. CYPs 3A4, 3A5, and 2C9 are the predominant intestinal isoforms, accounting for approximately 80% (3As) and 15% (2C9) of the total, respectively, with the remainder comprising CYPs 1A1, 1A2, 2C19, 2J2, and 2D6 (Paine et al., 2006). CYP3A expression declines from the proximal to distal intestine, with 75% occurring in the duodenum and jejunum (Paine et al., 1997). In the intestine, like in the liver, CYP3A4 is the predominant CYP3A isoform, and CYP3A5 is polymorphically expressed in 20% to 70% of adults (Paine et al., 1997). In otherwise histologically normal duodenal biopsies from a population of 74 children, CYP3A4 protein expression increased steadily with age (Fig. 7-7) (Johnson and Thomson, 2008). In fetal duodenum it was essentially absent, and in neonates it was expressed at about half the level seen in mature children. Intestinal CYP3A enzyme activity followed the same pattern as enzyme expression (Johnson and Thomson, 2008).
Other phase I enzymes are important in drug metabolism. Ester hydrolysis is a ubiquitous reaction catalyzed by a diverse array of esterases in blood and tissue. Esterase activity is important in the hydrolysis of remifentanil to an inactive metabolite, resulting in termination of clinical effect. Remifentanil is hydrolyzed by nonspecific esterases in plasma and (more so) tissue, but not by plasma cholinesterase (Manullang and Egan, 1999). In contrast, and unlike most ester drugs, red-cell rather than plasma esterases metabolize esmolol. Whereas CYPs mostly mature in the first months to year of life, esterase activity in neonates is already at levels nearly equivalent to those in adults (Allegaert et al., 2008). For example, remifentanil clearance in children from the ages of 1 month to 9 years old resembles that in adults (Sumpter and Anderson, 2009).
Phase II drug-metabolizing enzymes include glucuronosyltransferases (UGTs), sulfotransferases (SULTs), acetyltransferases, and glutathione transferases, all catalyzing conjugation reactions with multiple isoforms of each enzyme, often with isoform-dependent ontogeny (Blake et al., 2005; Hines, 2008). The two main UGT families are UGT1 and UGT2. UGT1A1, the major enzyme responsible for bilirubin conjugation, is not detectable in fetal liver, increases immediately after birth, and reaches adult levels by 3 to 6 months of age. UGT2B7 is of particular interest, because it conjugates morphine. UGT2B7 activity in fetal liver is present at 10% to 20% of adult values and increases in neonates to reach adult values by 2 to 3 months after birth. Morphine clearance is very low in premature infants and increases substantially over the first year of life. SULT ontogeny is isoform-specific, with SULT1E1 activity highest in the fetus and declining thereafter, SULT2A1 activity is barely detectable in neonates and increases in neonates, whereas SULT1A1 activity is constant from fetal age to adulthood.
Elimination
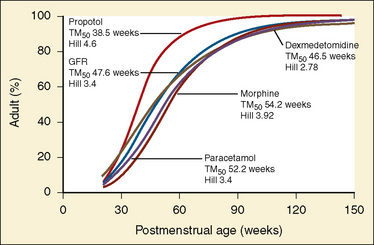
Elimination refers to all processes that remove a drug from the body, including drug metabolism (biotransformation) and excretion. Returning to the concept of drug elimination in general, it is now appreciated that several elements affect the development of drug elimination from fetus to adult, including patient size, organ maturation, organ function, and coexisting disease. Developmental patterns of drug clearance are shown for several drugs in Figure 7-8. For drugs in which renal elimination largely determines systemic clearance, the age-dependence of such clearance approximates that of GFR. In contrast, when hepatic metabolism predominates, the pattern differs.
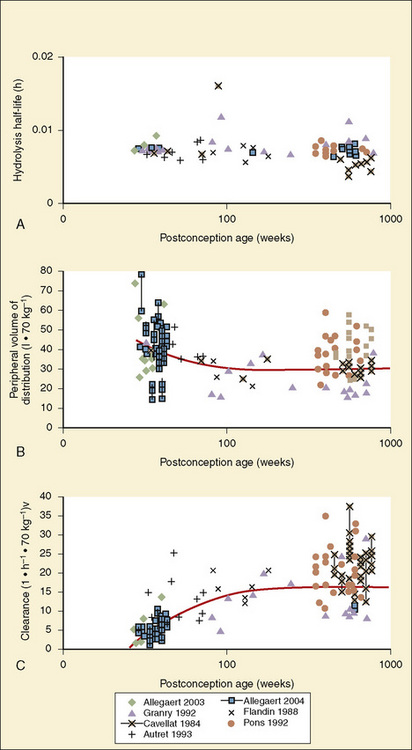
The complexity of developmental pharmacokinetics can be appreciated using a drug such as propacetamol. Propacetamol is an N, N-diethylglycine ester prodrug of acetaminophen (paracetamol), which is hydrolyzed by plasma esterases after intravenous administration to the active metabolite acetaminophen. Acetaminophen in turn undergoes both phase I, and more so phase II metabolism, to glucuronic acid, sulfate, cysteine, and glutathione conjugates. Figure 7-9 shows the developmental aspects of propacetamol pharmacokinetics (Anderson et al., 2005). Consistent with the relative age invariance of esterase expression, ester hydrolysis to the active metabolite, acetaminophen, was also age invariant. The central compartment volume of distribution was also age invariant, whereas the peripheral compartment volume was somewhat decreased in neonates. Clearance increased markedly with age in the first year, from 12% of adult values at 27 weeks’ PCA to 84% of its mature value by 1 year. Importantly, PCA was more important than PNA, as described above.
Scaling Pediatric Dosing
Ideal clinical practice would be informed by age-specific clinical pharmacokinetic data for every drug used in children. As this remains an unattained ideal, considerable effort has been expended to establish pharmacokinetic models to predict age-dependent dosing based on adult data (Anderson and Holford, 2008; Johnson, 2008 Tod et al., 2008; Sumpter and Anderson, 2009). Several principles are now apparent, some of which have been described previously in this chapter. Loading doses depend on concentration and central volume of distribution. For central volumes, allometric modeling (p. 195) suggests that the volume of distribution scales with a power of 1, such that
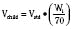
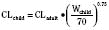
Figure 7-10 compares the accuracy of the linear per-kilogram, the body-surface area, and the allometric three-fourths power models.
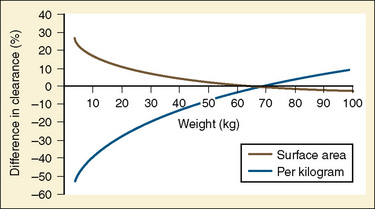
Clearance, however, depends on not just growth (size), but also on maturation of organ clearance processes (i.e., PCA). It is also influenced by concomitant diseases and potentially by drug interactions, which can influence organ function. While allometry alone provides reasonable estimates of clearance in older children using adult data, it alone is insufficient to predict clearance in infants and young children (Sumpter and Anderson, 2009). Therefore, the most parsimonious model for prediction drug clearance is as follows:
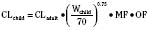
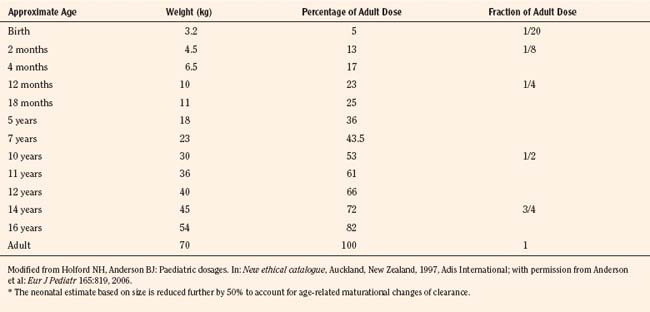
For practical purposes, size models have been simplified for clinical use. This is also influenced by regulatory considerations, which sort children into the categories of neonates (younger than 1 month), infants (1 month to 2 years), children (2 to 12 years), and adolescents (12 to 18 years). Table 7-5 presents age-specific dose adjustments. An even simpler method suggests 1 month, 1 year, 7 years, and 12 years, respectively for doses that are one eighth, one fourth, one half, and three fourths of the adult doses (Anderson and Holford, 2008). It is important to remember that these dose adjustments are for drug disposition only, and they do not take into account any age-dependent differences in pharmacodynamic response.
Pharmacodynamics
True age-related pharmacodynamic changes may be qualitative or quantitative, and they may apply to both therapeutic and adverse effects (Stephenson, 2005). One example of quantitative differences in therapeutic response is the effect of warfarin in prepubertal, pubertal, and adult patients. Despite equivalent plasma drug concentrations, warfarin effects on prothrombin fragments 1 and 2 and on the international normalized ratio (INR) were higher in prepubertal patients than in adults. Similarly, augmented response was observed with cyclosporine, in which peripheral blood monocytes from infants had twofold lower proliferation and sevenfold lower interleukin-2 expression compared with older subjects. Examples of age-dependent adverse effects occurring only in children include chloramphenicol toxicity (“gray baby syndrome”), limb deformation from thalidomide during embryogenesis, tetracycline staining of dental enamel, and valproic acid hepatotoxicity, which is increased in young children. Generalized factors leading to age-dependent drug responses include physiology, pathology, host response to disease, and adverse drug reactions, as well as pharmacodynamics (Stephenson, 2005).
For drugs used in anesthesia, the best information on developmental pharmacodynamics has been obtained for inhaled anesthetics, and to a lesser extent, certain intravenous anesthetics. The potency of inhaled anesthetics is significantly affected by developmental age. The principal metric of inhaled anesthetic potency (the median effective concentration, or EC50), has been called the minimum alveolar concentration (MAC) and defined as the “minimum alveolar concentration of anesthetic at 1 atmosphere that produces immobility in 50% of those patients or animals exposed to a noxious stimulus,” where the stimulus is usually an incision (Mapleson, 1996). Other endpoints have been analogously defined, such as MACAWAKE endpoint, which defines loss (or return) of consciousness. The quantal EC50 of all inhaled anesthetics is similarly influenced by age, with a common log-linear negative slope (for age older than 1 year) (Mapleson, 1996). For decreasing ages younger than 40 years, EC50 (MAC) increases 6% per decade. Thus, the MAC for sevoflurane is conceptually 20% greater at age 10 and 27% greater at age 1 (i.e., 2.16% and 2.29% at ages 10 and 1, respectively, for a MAC of 1.8% at age 40 (Mapleson, 1996). A clinical study found the MAC of sevoflurane to be 2.5% in children between 1 and 12 years old and 3.2% in infants 6 to 12 months old, compared with 2% at age 40 (Lerman et al., 1994; Mapleson, 1996). The MACAWAKE endpoint for sevoflurane was 0.43%, 0.45%, and 0.66% in children 8 to 12 years old, 5 to younger than 8 years, and 2 to younger than 5 years, respectively (Davidson et al., 2008c). The ratio between the MACAWAKE endpoint and MAC did not differ with age.
Measures of the age-dependent change in apparent anesthetic potency may be influenced by the clinical drug effect used as the index of response. For example, various electroencephalogram (EEG)-derived parameters (e.g., spectral edge frequency, bispectrum, and bispectral index [BIS]) have been used to measure volatile anesthetic effects in children (Wodey et al., 2005; Tirel et al., 2006; Davidson et al., 2008b). Although there were age-dependent effects of volatile anesthetics on various EEG parameters, the EEG itself was found to be highly age-dependent. Specifically, EEG was fundamentally different in infants between 0 and 6 months old, and caution was suggested in the use of BIS to determine volatile anesthetic pharmacodynamics in children (Wodey et al., 2005; Davidson et al., 2008b). Similar results were obtained when EEG-derived parameters were used to evaluate age and propofol effects, and EEG results in children younger than 1 year old were considered inaccurate (Jeleazcov et al., 2007).
Compared with inhaled anesthetics, much less is known about developmental aspects of intravenous drug pharmacodynamics. In part, this reflects the ease, lack of expense, ubiquity, and real-time availability of measuring end-tidal inhaled anesthetic concentrations, compared with measuring plasma concentrations of intravenous anesthetics. Limited data are available for propofol, which suggests slightly lower sensitivity (diminished potency) in children. Plasma propofol concentration-effect (BIS) curves analyzed in children (mean age of 10 years, range of 6 to 13 years) and adults (mean age of 18 years, range of 14 to 32 years) found graded EC50 means values of 4 vs. 3.3 mcg/mL, respectively. When propofol infusions were targeted to maintain a steady-state BIS of 50, measured mean plasma concentrations were 4.3 ± 1.1 and 3.4 ± 1.2 mcg/mL, respectively (Rigouzzo et al., 2008). Somewhat lower propofol potency was also reported by others (Jeleazcov et al., 2008). In contrast, one study found no difference in propofol EC50 in adults and children, however, predicted rather than measured plasma concentrations were used in the analysis, which is a limitation of such studies (Munoz et al., 2006). Increased propofol EC50 in children is consistent with diminished EC50 in older adults (Schnider et al., 1999). No data are available regarding propofol pharmacodynamics in neonates.
Nonlinear Pharmacokinetics
The nonlinear pharmacokinetics may also be seen in low-clearance drugs for which elimination is significantly influenced by the binding of the drug to plasma proteins. In this scenario, after increasing the dose of the drug, a less-than-expected increase in Css and AUC occurs. This would suggest that the plasma protein-binding sites have been saturated and that the free fraction of low-clearance drug has increased. The latter would result with increased clearance and a less-than-expected increase Css occurs. However, if measured, the free fraction of the low-clearance drug increases proportionally. Both valproic acid and disopyramide follow this type of nonlinear pharmacokinetics (Bowdle et al., 1980; Lima et al., 1991).
Compartment Models
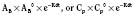
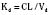
The elimination rate constant (Kd) can be also thought of as the fraction of the volume of distribution that is effectively cleared of drug per unit of time. Because the drug plasma concentration diminishes monoexponentially, a graph plot of the logarithm of the plasma concentrations vs. time yields a straight line. The elimination rate constant defines the slope of this curve, and two plasma concentrations measured during the decay or elimination phase can be used to calculate the Kd (Fig. 7-11):
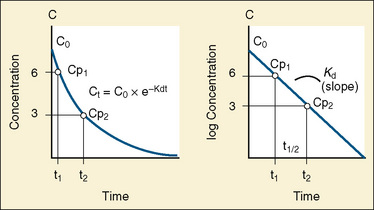
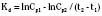
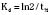
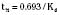
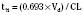
The half-life is a variable that determines the following factors:
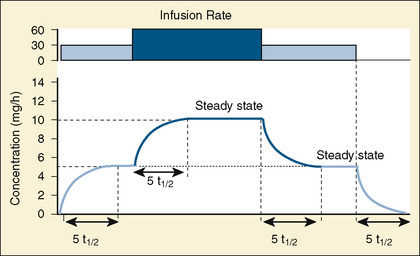
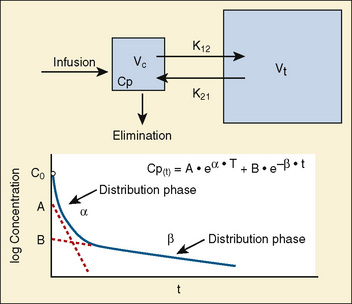
Most of the drugs used in anesthesia do not follow the simple, one-compartment pharmacokinetics but rather behave like a two- or even three-compartment model. Distribution of anesthetic drugs into and out of peripheral tissues determines the pharmacokinetic profile and the time course of the anesthetic drug’s effect. For the two-compartment model, the central compartment includes the blood and organs or tissues that have high blood flow and can be thought of as a rapidly equilibrating volume. The second compartment has a volume (Vt) that equilibrates at a much slower pace. After bolus administration of a drug that follows the two-compartment model, the two distinct phases of distribution and terminal elimination can be distinguished, and the decay of plasma concentration over time is defined by the biexponential equation (Fig. 7-13). Changes in plasma and the site-of-action concentrations would depend on drug elimination and on the equilibrium between central and peripheral tissue compartments.
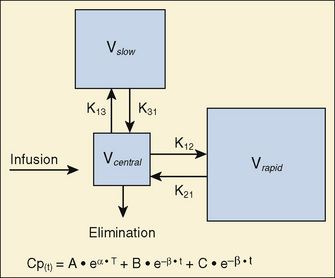
For many anesthetic drugs, three phases can be distinguished after intravenous bolus administration. This three-compartment model is composed of the central compartment and two additional compartments that include the respective rapid and slow equilibrating tissues and organs (Fig. 7-14). Likewise, the three-compartment model is characterized by the triexponential plasma concentration equation, three volumes of distribution, and five rate constants of distribution and terminal elimination (K12, K21, K13, K31, and K10).
Context-Sensitive Half-Time
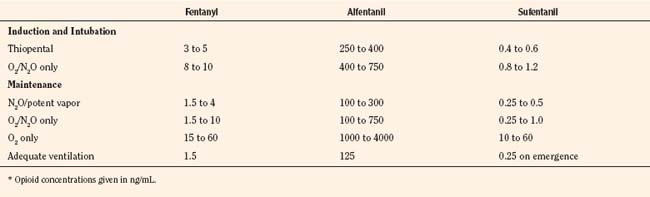
For the anesthesiologist, pharmacokinetic factors are often used for the routine selection and use of various intravenous anesthetic agents. Drugs with short elimination half-lives are commonly selected for brief procedures, whereas drugs with longer half-lives are selected for lengthier procedures. Drugs with small volumes of distribution tend to decrease the time required for recovery after intravenous infusion, and agents with decreased plasma clearances may increase the time for recovery. In general, formulas for the calculation of continuous infusions incorporate knowledge of these pharmacokinetics. For bolus administration of drug, the volume of distribution and the desired plasma concentration are needed. Table 7-6 lists plasma concentrations in adults for some of the opioids. The bolus dose is calculated as the product of the volume of distribution and the desired plasma concentration (Cp):
TABLE 7-6 Opioid Concentrations that Ablate Responsiveness to Intraoperative Noxious Stimuli and Permit Adequate Ventilation on Emergence*
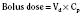
Although these formulas work well, Shafer and Varvel (1991) and Hughes et al. (1992) have demonstrated the complex interactions that occur with prolonged infusions, especially in drugs that are lipid soluble.
The offset of drug effect depends on reduction of the plasma concentrations and the withdrawal of drug from the site of action; that is, for anesthetic from the receptor site in the CNS. If steady state is achieved and all compartments are saturated, then the half-life of elimination phase, which is a function of the first-order processes of elimination, correlates with a decrease in the site-of-action drug concentrations and with the offset of drug action. However, after infusion of a highly lipophilic anesthetic agent, when steady state is not achieved and not all compartments are saturated, the decline in concentrations (i.e., the offset of action and recovery from anesthesia) depends on complex interactions between the duration of the infusion and initial distribution, redistribution, and metabolic and elimination first-order processes. The classic descriptors of a drug’s pharmacokinetics and offset of action (terminal half-time) are of little help to anesthesiologists in predicting the offset of action and recovery from anesthesia for the intravenous anesthetic drugs. Fortunately, with the help of pharmacokinetic and pharmacodynamic simulation models, new predictors of offset of drug effects have evolved (Shafer and Varvel, 1991; Hughes et al., 1992; Youngs and Shafer, 1994).
Using pharmacokinetic and pharmacodynamic models and basic pharmacokinetic profiles of commonly used synthetic opioid analogs, Shafer and Varvel (1991) were the first to construct the offset of action (recovery) curves as a function of the duration of infusion for fentanyl, alfentanil, and sufentanil (Fig. 7-15). Hughes et al. (1992) introduced the term context- sensitive half-time (context refers to the duration of infusion) as a time required for a drug concentration to decrease to half of its value after drug infusion of a given duration. Of importance is that for any given drug, its context-sensitive half-time varies with the duration of the drug infusion. Because for most intravenous anesthetic drugs the 50% fall in concentration is not sufficient for recovery from anesthesia, other decrement times have been introduced (Youngs and Shafer, 1994), for example, 80% and 90% decrements.
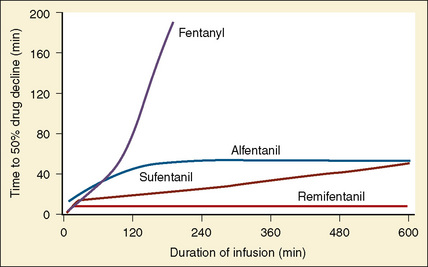
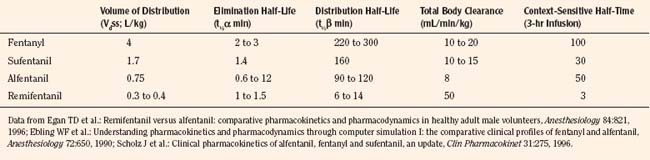
After a short intravenous infusion (0 to 15 minutes), fentanyl and other synthetic opioid analogs have similar context-sensitive half-times (Fig. 7-15). However, after prolonged infusion (longer than 1 hour), there is a marked difference among four synthetic opioid analogs (fentanyl, sufentanil, alfentanil, and remifentanil), and these differences do not correlate with their classic pharmacokinetic parameters (Table 7-7). In contrast to its older congeners, remifentanil has a short and steady context-sensitive half-life of 3 minutes, which does not change with the increasing duration of infusion (Egan et al., 1996). This contrasts with alfentanil, which has a context-sensitive half-time that increases to 1 hour after a 4-hour infusion (Ebling et al., 1990; Scholz et al., 1996). This difference is because of the unique pharmacokinetic profile of remifentanil. It is a highly liposoluble (volume of distribution at steady state [Vdss] of 30 L) opioid analog that undergoes widespread metabolism (deesterification), including metabolism in the circulation. Unlike other opioids, the termination of action of remifentanil does not depend on redistribution but rather on extremely rapid metabolic clearance. Kapila et al. (1995) demonstrated that the context-sensitive half-times of remifentanil (3 minutes) and alfentanil (50 to 55 minutes) derived from computer modeling are similar to measured context-sensitive half-times (3.2 and 47 minutes for remifentanil and alfentanil, respectively), and both correlate with measured pharmacodynamic offset (recovery of minute ventilation). The latter confirms the value and clinical applicability of this new pharmacokinetic-pharmacodynamic parameter in predicting the offset of anesthetic drugs.
TABLE 7-7 Pharmacokinetic Parameters of Synthetic Opioids after Single Intravenous Bolus Administration
The context-sensitive half-time curves provide a better, clinically more relevant comparison of the pharmacokinetic profiles of anesthetic drugs than the traditional pharmacokinetic parameters (Fig. 7-16). After a single intravenous dose, commonly used anesthetic and hypnotic drugs have a short duration of action. However, after prolonged infusions the context-sensitive half-times and duration of action increase. For some drugs (e.g., propofol and ketamine), this increase is modest, whereas for others (e.g., diazepam and thiopental), it is quite dramatic. In the case of midazolam, the rapid increase in its context-sensitive half-time with prolonged duration of infusion occurs in the presence of a relatively short elimination half-life (t½β), and this is most likely because of the low clearance of midazolam.
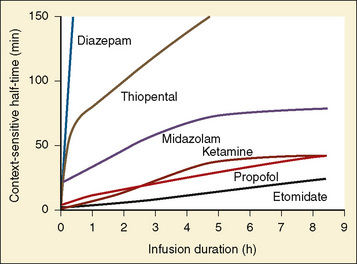
FIGURE 7-16 Context-sensitive half-time for commonly used general anesthetics.
(From Reves JG et al.: Nonbarbiturate intravenous anesthetics. In Miller RD, editor: Anesthesia, ed 5, New York, 2000, Churchill Livingstone.)
The data regarding the context-sensitive half-times and other decrement times of anesthetic agents in the pediatric population are limited, at best. In children aged 3 to 11 years, longer context-sensitive half-times than in adults were reported for propofol. After a 1-hour infusion in children and adults, the context-sensitive half-times for propofol are 10.4 and 6.6 minutes, respectively, and after a 4-hour infusion, they are 19.6 and 9.5 minutes, respectively (McFarlan et al., 1999). This is probably due to the altered compartment volumes and may lead to slower recovery from a propofol infusion in children than in adults (Short et al., 1994). In contrast, the shorter context-sensitive half-time of fentanyl was determined in pediatric population (2 to 11 years old) compared with published data in adults (Ginsberg et al., 1996).
Population Pharmacokinetics
Pharmacokinetics and pharmacodynamics of drugs in the pediatric population and in adults are different. Furthermore, neonates, infants, children, and adolescents have distinct differences in physiologic development, and the pediatric population is quite heterogeneous with regard to pharmacokinetics and pharmacodynamics of drugs across different age groups. These differences justify studying pharmacokinetics and pharmacodynamics in children of various ages. However, the testing of medications in children presents a dilemma: while society wants to spare children from the potential risk involved in research, children may be harmed if they are given medications that have been inadequately studied (Steinbrook, 2002).
PPK is an area of clinical pharmacology that studies the sources and the correlates of variability in drug-plasma concentration parameters among individuals in the target population of patients who are receiving clinically effective doses of a drug (Shen and Lu, 2007). PPK is focused on the quantitative assessment of the typical pharmacokinetic parameters, as well as the within- and between-individual and residual variability in drug absorption, distribution, metabolism, and excretion (Steiner, 1992; Ette and Williams, 2004). The population approaches use mathematic and statistic modeling to investigate the dose-concentration-effect relationship and to quantitatively and qualitatively assess factors that may explain interindividual variability (Sheiner et al., 1979). In the early 1980s, Sheiner and Beal introduced the new PPK data analysis approach (i.e., the population approach) and demonstrated that estimates of PPK parameters could be obtained even if only two or three samples are collected per patient (Sheiner and Beal, 1980, 1981, 1982). They also introduced a new software program (the Nonlinear Mixed Effects Model, or NONMEM) that was capable of performing the new type of analysis, and this is now the most commonly used population modeling program (Sheiner and Beal, 1980; Sheiner et al., 1979).
The PPK approach allows not only major contributions to variability of key pharmacokinetic parameters to be established, but it also goes one step further and allows the relative contributions of different factors to be determined. In this regard, size, age, and renal function are major contributors to vancomycin clearance variability in neonates. Using the PPK and NONMEM modeling, Anderson and colleagues (2002a) have shown that size explains 49.8%, age accounts for 18.2%, and renal function explains 14.1% of clearance variability of vancomycin in neonates. The small unexplained percentage (18%) is residual variability in clearance and suggests that target concentration intervention is unnecessary if size, age, and renal function are used to predict the dose (Anderson et al., 2002a).









