Fig. 1
Hyperalgesic priming induces a switch in nociceptor second messenger signaling pathways. In a naïve animal, administration (hindpaw injection) of an inflammatory stimulus (e.g., PGE2) results in transient Gs/PKA-mediated acute pain (green curve in (a) and left side in (b)). In the primed animal, the same inflammatory stimulus recruits an additional Gi/o/PKCε and/or an Epac/PKCε-mediated pathway, which contributes to prolonged hyperalgesia (red curve in (a) and right side in (b)) in previously primed animals. Adapted from Reichling and Levine (2009)
3.1 PKCε as a Crucial Mechanism of Nociceptor Priming
How then is this exaggerated response to PGE2, and other mediators like serotonin or A2 adenosine receptor agonists (Aley et al. 2000), which can also precipitate hyperalgesia in primed animals, generated? Extensive studies have demonstrated that this priming effect is dependent on switches in signaling mechanisms in nociceptors. In naïve animals, the hyperalgesic effects of PGE2 injection are mediated by adenylyl cyclase (AC) activation downstream of PGE2 receptors causing protein kinase A (PKA) activation (Fig. 1b) (Aley and Levine 1999). This effect can be attenuated via injection of PKA antagonists (Aley and Levine 1999). Although the second messenger pathway underlying the early phase of PGE2-induced hyperalgesia is PKA mediated, even in primed animals, the same cannot be said for the pathway responsible for the long-lasting hyperalgesia that is uniquely present in primed rodents. While PGE2-induced hyperalgesia in primed animals is still cyclic AMP (cAMP) dependent, it now bypasses PKA to activate exchange proteins directly activated by cAMP (Epac) which can activate protein kinase Cε (PKCε, Fig. 1b, Hucho et al. 2005). Importantly, inflammatory stimulation of nociceptors leads to a decrease in G-protein receptor kinase 2 (GRK2) which results in enhanced Epac activity (Eijkelkamp et al. 2010; Wang et al. 2013). These changes occur in IB4-positive nociceptors, and decreases in GRK2 and increases in Epac expression are correlated with the persistence of priming (Wang et al. 2013). Moreover, in primed animals, PGE2 results in an activation of pertussis toxin-sensitive G-protein αi subunits (Dina et al. 2009) and phospholipase Cβ (PLCβ) leading to a downstream engagement of PKCε (Joseph et al. 2007); hence, multiple pathways for PKCε engagement may exist in primed nociceptors (Fig. 1b). Critically, in primed animals, the long-lasting hyperalgesia arising from exposure to compounds that can precipitate priming is blocked by selective inhibition of PKCε (Aley et al. 2000) and by intrathecal delivery of antisense oligonucleotides knocking down PKCε expression (Parada et al. 2003). Additionally, injection of a PKCε agonist alone results in a prolonged hyperalgesic state and hyperalgesic priming, pointing to a key role for nociceptor PKCε in hyperalgesic priming (Reichling and Levine 2009). Importantly, this PKCε-dependent primed state does not require an initial bout of hyperalgesia as subthreshold doses of PKCε activators (Parada et al. 2003), previous exposure to unpredictable sound stress (Khasar et al. 2008), and even repeated administration of opioid agonists into the paw (Joseph et al. 2010) are capable of causing an emergence of a primed state (Fig. 1a).
Is there a distinct subset of nociceptors required for PKCε-dependent priming? To elucidate the population of nociceptors involved in hyperalgesic priming, Joseph and colleagues lesioned IB4(+) nociceptors via intrathecal administration of IB4-saporin (Joseph and Levine 2010). In these animals, the PKCε activator, ψεRACK, produces an initial hyperalgesia; however, the PGE2-induced prolonged hyperalgesia is absent in animals treated with toxin suggesting that priming is localized to IB4(+) neurons. Furthermore, select agents known to act on peptidergic vs. IB4(+) cells stimulate an initial hyperalgesic state in IB4-lesioned animals but fail to establish priming indicating that a switch in signaling to PKCε selectively occurs in IB4(+) nociceptors (Joseph and Levine 2010). As mentioned above, this may be linked to a pronounced loss of GRK2 expression and an increase in Epac activity in IB4(+) nociceptors of primed animals (Wang et al. 2013).
Collectively these findings support a key role for PKCε in the initiation and maintenance of nociceptor priming therefore making this PKC isoform a key therapeutic target. However, PKCε-dependent forms of priming show a marked sexual dimorphism in rats with female rats failing to show priming, an effect that is apparently due to a protective effect of estrogen (Joseph et al. 2003). Importantly, this does not appear to be the case with mice where both carrageenan and PKCε-activating peptides cause robust priming to subsequent PGE2 injection in females (Wang et al. 2013). At this point, it remains unclear as to whether there is sexual dimorphism in PKCε-mediated effects in humans. Despite these potential discrepancies, it is clear that one mechanism of priming is shared across species and sexes, a requirement for changes in gene expression at the level of protein synthesis.
3.2 Local Translation Is a Key Mediator of Nociceptor Priming
Due to the continuous turnover of cellular proteins, a simple switch in G-protein coupling is likely not sufficient to explain such a long-lasting maladaptive change in peripheral nociceptors (Bogen et al. 2012). It is likely that this effect is coupled to changes in gene expression and a possible solution to this problem is a localized change occurring at the level of protein translation. Translation can be controlled by extracellular factors signaling via kinase cascades offering a rapid, locally mediated control of gene expression. Two important kinases for translation control are the mechanistic target of rapamycin complex 1 (mTORC1) and extracellular signal-regulated kinase (ERK) (Fig. 2a). Both of these kinases signal to proteins that bind to the 5′ cap structure of mRNAs. mTORC1 phosphorylates 4E-binding proteins (4EBPs) leading to a release of inhibition on eukaryotic initiation factor (eIF) proteins allowing for the eIF4F complex (composed of eIF4E, the 5′ cap-binding protein; eIF4G, a scaffolding protein; and eIF4A, a helicase that unwinds secondary structure in mRNAs) to form (Fig. 2a) (Sonenberg and Hinnebusch 2009). ERK, on the other hand, activates the mitogen-activated kinase-interacting kinases (MNKs), which then phosphorylate eIF4E (Fig. 2a) (Wang et al. 1998). Phosphorylation of eIF4E, which is mediated specifically by MNK1/2, is thought to enhance eIF4F complex formation leading to an increase in translation (Ueda et al. 2010; Herdy et al. 2012). In sensory neurons, nerve growth factor (NGF) and interleukin 6 (IL6), two factors known to induce priming, induce an increase in ERK and mTORC1 signaling leading to a local, axonal increase in protein synthesis (Melemedjian et al. 2010, 2014). Blockade of these kinases, or blockade of eIF4F complex formation with the eIF4F inhibitor compound 4EGI1 (Fig. 2a), inhibits mechanical hypersensitivity induced by these factors and abrogates priming to subsequent PGE2 exposure (Melemedjian et al. 2010; Asiedu et al. 2011). Hence, local translation is required for the induction of priming downstream of NGF and IL6 signaling.
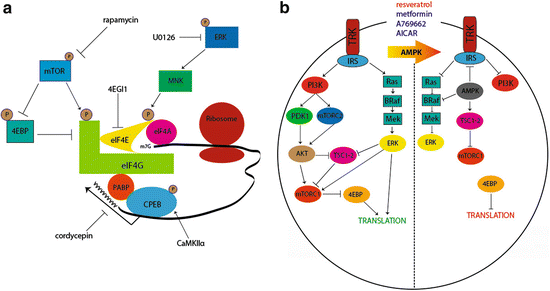
Fig. 2
Translational control pathways involved in hyperalgesic priming. (a) mTORC1 phosphorylates 4EBPs, negative regulators of eIF4F formation. This results in its dissociation from eIF4E, allowing the binding of eIF4E to eIF4G. Phosphorylation of eIF4E (via ERK/MNK1/2) or eIF4G (via mTORC1) enhances the formation of the eIF4F complex, promoting translation. Phosphorylation of CPEB by CamKIIα enhances translation efficiency by increasing the length of the poly A tail in mRNAs containing a CPE sequence. Taken together, eIF4F cap complex formation enhances cap-dependent translation, which is necessary for the induction of priming via translational control of gene expression in sensory afferents. (b) Pharmacological activation of AMPK results in the inhibition of IRS-1, dampening Trk-mediated signaling. AMPK activation also results in TSC2 phosphorylation, thereby inhibiting mTORC1. Moreover, AMPK phosphorylates BRaf leading to inhibition of ERK signaling. Taken together, AMPK activation decreases activity-dependent translation by turning off both the ERK and mTORC1 pathways, presenting a novel opportunity to prevent or reverse hyperalgesic priming. Adapted from Melemedjian et al. (2010) and Price and Dussor (2013)
An alternative mechanism to decrease ERK and mTORC1 signaling is via stimulation of adenosine monophosphate-activated protein kinase (AMPK) (Fig. 2b). AMPK is a ubiquitously expressed energy sensing kinase well known to inhibit mTORC1 signaling through multiple phosphorylation events (Carling et al. 2012). AMPK also abrogates ERK signaling in many cell types, and this effect has recently been linked to negative regulation of the upstream ERK activator BRaf (Fig. 2b) (Shen et al. 2013). In sensory neurons, AMPK activation with pharmacological stimulators (for review see Price and Dussor 2013) leads to decreased ERK and mTORC1 activity (Melemedjian et al. 2011; Tillu et al. 2012), decreased eIF4F complex formation (Melemedjian et al. 2011; Tillu et al. 2012), and inhibition of axonal protein synthesis as measured by enhanced processing body (P body) formation (Melemedjian et al. 2014). AMPK activators decrease peripheral nerve injury- and inflammation-induced mechanical hyperalgesia (Melemedjian et al. 2011; Russe et al. 2013) suggesting an important role for this kinase in pain plasticity. In the context of hyperalgesic priming, AMPK activation with the natural product resveratrol (Fig. 2b) decreases mechanical hypersensitivity caused by incision and completely blocks the development of priming when given locally around the time of incision (Tillu et al. 2012). These findings further suggest a role for local translation in the initiation of plasticity leading to priming of nociceptors.
Epac signaling, which was described above as an important mediator of priming induced by inflammatory mediators (Wang et al. 2013), may also play an important role in regulating translation in sensory neurons. Decreased GRK2 leads to enhanced activation of Epac causing an increase in ERK activity (Eijkelkamp et al. 2010). While this signaling event has not been linked to translation control, based on findings involving IL6-induced priming and its dependence of ERK/eIF4E-mediated changes in translation control, it is conceivable that this is an important mediator for priming where ERK activity is increased. Furthermore, Epac activation causes an increase in mTORC1 activity in some transformed cells (Misra and Pizzo 2009) suggesting that enhanced Epac signaling in primed nociceptors may lead to convergent signaling onto the eIF4F complex in a manner analogous to that observed with NGF and IL6 (Melemedjian et al. 2010). It remains to be seen if these Epac-mediated events occur in sensory neurons and their axons, but this may be a fruitful area for further work and therapeutic intervention.
The regulation of translation via 5′ cap-binding proteins and their upstream kinases clearly comprise an important mechanism for the priming of nociceptors. However, translation is likewise regulated by RNA-binding proteins that bind to either 5′ or 3′ untranslated regions (UTRs). The fragile X mental retardation protein (FMRP) is a key RNA-binding protein regulating plasticity in the PNS and CNS (Bassell and Warren 2008). Mice lacking FMRP fail to sensitize in several preclinical pain models (Price et al. 2007; Price and Melemedjian 2012), and these mice also have deficits in priming induced by NGF and IL6 (Asiedu et al. 2011). Another important RNA-binding protein in hyperalgesic priming is the cytoplasmic polyadenylation element-binding protein (CPEB). CPEB binds preferentially to mRNAs containing a CPE sequence in their 3′ UTR near the polyadenylation sequence. These mRNAs contain short poly A tails, and CPEB, upon phosphorylation, can enhance the poly A tail length leading to enhanced translation efficiency (Fig. 2a) (Richter 2007). This process is linked to long-term potentiation (LTP) in the CNS (Udagawa et al. 2012) and has recently been shown to play an important role in nociceptor priming (Bogen et al. 2012; Ferrari et al. 2012, 2013a). CPEB is phosphorylated by the aurora family kinases (Mendez et al. 2000) and, likely more importantly for neurons, by Ca2+/calmodulin-activated protein kinase IIα (Fig. 2a) (CaMKIIα, Atkins et al. 2005). In the PNS, CPEB is primarily expressed by IB4(+) nociceptors (Bogen et al. 2012). Knockdown of CPEB with intrathecally injected antisense oligonucleotides leads to inhibition of the initiation of priming induced by PKCε activators linking translation events to PKCε-dependent priming (Bogen et al. 2012). Importantly, in priming induced by peripheral inflammation, CPEB may act downstream of PKCε and CaMKIIα to initiate and maintain a primed state (Ferrari et al. 2013a). Since CPEB is thought to have prion-like properties that are crucial for its role in memory maintenance (Si et al. 2003a, b), these findings point to a potential role for CPEB in creating a permanently primed state in peripheral nociceptors.
While it is clear that translation regulation is required to initiate a primed state in the periphery, a crucial question is whether disruption of local translation is capable of interrupting priming once it has been fully established. This is an important question because it gives insight into devising therapeutic strategies to reverse priming in the clinical state. While there are certain situations (e.g., surgery) where inhibition of priming mechanisms at the time of injury is a viable strategy, the majority of clinical situations are likely to require intervention following the full establishment of priming. An experimental paradigm to test this translation dependency is to induce priming with a locally administered stimulus and then allow the initial hyperalgesia to resolve. Then, prior to injection of the stimulus to precipitate hyperalgesia in primed animals, translation inhibitors can be administered locally to test whether continuous translation is required to maintain a primed state (Asiedu et al. 2011; Ferrari et al. 2013a). In this regard, following injection of IL6 and resolution of mechanical hyperalgesia in mice, injection of anisomycin or rapamycin (at doses that block the initiation of priming) fail to reverse a primed state (Asiedu et al. 2011). In contrast, in rats, injection of carrageenan causes priming that is disrupted both at the time of carrageenan injection and during the maintenance phase by either the mTORC1 inhibitor rapamycin or the polyadenylation inhibitor cordycepin (Ferrari et al. 2013b). Interestingly, in addition to cordycepin action on polyadenylation, this compound has recently been identified as an AMPK activator (Wu et al. 2014). Similar effects with rapamycin and cordycepin are observed in rats primed with paw injection of CaMKIIα. Since CaMKIIα phosphorylates CPEB and CPEB regulates CaMKIIα translation, this raises the intriguing possibility that CaMKIIα/CPEB signaling could represent a positive feedback mechanism to maintain pain memory in the peripheral nociceptor (Ferrari et al. 2013a). Importantly, in rats where PKCε-induced priming is sexually dimorphic, priming that is dependent on local translation occurs both in male and female rats (Ferrari et al. 2013a). Hence, while there are conflicting results in different models, it is formally possible that brief disruption of local translation in primed nociceptors is capable of resolving a primed state. Further work is clearly needed to further interrogate mechanisms involved in these effects; however, they point to the tantalizing possibility that therapeutics targeting translation regulation might have disease-modifying effects in chronic pain conditions. This hypothesis is consistent with the observation that AMPK activators, which have a strong effect on translation regulation pathways, have disease-modifying effects in neuropathic pain models (Melemedjian et al. 2011, 2013b).
4 CNS Regulation of Hyperalgesic Priming
4.1 Atypical PKCs and Brain-Derived Neurotropic Factor
While there is strong evidence for memory-like mechanisms controlling hyperalgesic priming at the level of the peripheral nociceptor, plasticity in the CNS, especially in the dorsal horn of the spinal cord, also plays a central role in hyperalgesic priming. Here, analogies to mechanisms of memory formation and maintenance in other CNS regions, such as the hippocampus and cortex, have played a central role in directing research in the area. LTP, long recognized as a neurophysiological correlate of learning and memory at central synapses, can be divided into an early and late phase. Early LTP requires the activation of CaMKIIα, PKA, and conventional PKC leading to the phosphorylation of AMPA receptors (Huganir and Nicoll 2013). Early LTP also leads to changes in gene expression which occur both on the level of transcription and translation. These changes in gene expression are needed for the consolidation of early LTP into late LTP (Abraham and Williams 2008). Mechanisms involved in the maintenance of late LTP have been more difficult to clearly elucidate but are thought to involve brain-derived neurotropic factor (BDNF) and an atypical PKC (aPKC) isoform called PKMζ (Fig. 3) (Sacktor 2011). While LTP has been extensively studied in the cortex and hippocampus, LTP can also be induced at central synapses in the dorsal horn of the spinal cord, and many mechanisms elucidated in other CNS regions are shared at these spinal synapses (Sandkuhler 2007). Spinal LTP is a potential mechanism for primary hyperalgesia (Sandkuhler 2007), and heterosynaptic LTP may explain other aspects of pain plasticity that occur in chronic pain disorders (Klein et al. 2008).
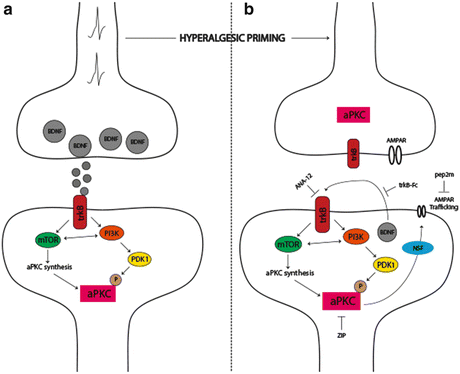
Fig. 3
The role of aPKCs in hyperalgesic priming initiation and maintenance. (a) Nociceptor activation leads to spinal BDNF release and a postsynaptic mTORC1-dependent translation of aPKC protein following trkB activation. These newly synthesized aPKCs are then phosphorylated by PDK1. Increased levels and phosphorylation of aPKCs are thought to be involved in initiating priming. (b) Once priming is established, increased aPKC protein and phosphorylation leads to a constitutive increase in AMPAR trafficking to the postsynaptic membrane. This appears to be regulated by BDNF signaling via trkB with BDNF potentially being released from postsynaptic dendrites in the maintenance stage of priming. Presynaptic trkB may also be activated by increased BDNF action in primed animals. Once established, hyperalgesic priming can be permanently reversed by inhibition of aPKCs with ZIP, by disruption of AMPAR trafficking with pep2M, or via inhibition of trkB/BDNF signaling with ANA-12 or trkB-Fc, respectively. Adapted from Price and Ghosh (2013)
Does spinal LTP explain features of the maintenance of hyperalgesic priming? While no direct measurements of spinal LTP as a correlate of hyperalgesic priming have been made, pharmacological similarities abound. Inhibition of translation and/or CaMKIIα during LTP induction blocks consolidation of late LTP (Nicoll and Roche 2013). Likewise, intrathecal injection of translation inhibitors or CaMKIIα inhibitors at the time of priming induction in mice inhibits initial hyperalgesia and prevents priming. In contrast, intrathecal injection of these compounds following the resolution of initial hyperalgesia fails to resolve priming consistent with a lack of effect of these mechanisms on the maintenance of late LTP (Asiedu et al. 2011; Melemedjian et al. 2013a). Late LTP can be reversed by inhibition of aPKCs with a pseudo substrate inhibitor called ZIP (Pastalkova et al. 2006). Intrathecal injection of ZIP either at the time of priming induction or following the resolution of the initial hyperalgesia leads to a complete reversal of hyperalgesic priming (Asiedu et al. 2011; Melemedjian et al. 2013a). ZIP also reverses established pain states that have become dependent on central plasticity following sustained afferent input (Laferriere et al. 2011). These findings are consistent with a role for PKMζ in the maintenance of late LTP, memory retention, and the maintenance of a chronic pain state. However, recent experiments using genetic methods to dissect the role of PKMζ in late LTP and memory maintenance have called the specificity of ZIP and the role of PKMζ in these effects into question (Lee et al. 2013; Volk et al. 2013). It remains to be seen if PKMζ plays a specific role in the maintenance of hyperalgesic priming in the dorsal horn of the spinal cord (for review on this topic see Price and Ghosh 2013).
An important component of the proposed role of PKMζ in LTP and memory is the trafficking of AMPA receptors to synaptic sites leading to a persistent augmentation of postsynaptic glutamate-mediated signaling (Fig. 3) (Sacktor 2011). This trafficking can be disrupted with a peptide called pep2m (Migues et al. 2010). Similar to experiments in other CNS regions, intrathecal injection of pep2m disrupts the maintenance of hyperalgesic priming (Asiedu et al. 2011) suggesting that aPKC-mediated regulation of AMPA receptor trafficking may play a central role in chronic pain states (Fig. 3). This is consistent with a wide variety of experimental findings indicating that AMPA receptor trafficking plays a central role in mediating pain plasticity induced by peripheral injury (Tao 2012).
As mentioned above, while it is clear that ZIP is capable of permanently reversing a primed state in a variety of experimental models (Asiedu et al. 2011; Laferriere et al. 2011; Melemedjian et al. 2013a), the target of ZIP is less clear based on recent evidence from transgenic mice (Lee et al. 2013; Volk et al. 2013). One possibility is that aPKC isoforms play a redundant role in synaptic plasticity, and therefore PKCλ may be involved in maintenance mechanisms of hyperalgesic priming (Price and Ghosh 2013). Since this isoform is also inhibited by ZIP (Melemedjian et al. 2013a; Volk et al. 2013), this is a parsimonious explanation for the discrepancy between pharmacological effects of ZIP and findings from mice lacking aPKCs derived from the Prckz locus (PKMζ and PKCζ). If this were the case, upstream mechanisms that regulate all aPKCs isoforms, either via phosphorylation or through their translation at synapses, would represent potential alternative targets to reverse hyperalgesic priming. A candidate molecule fitting this description is BDNF.
BDNF has long been recognized as an important mediator of pain plasticity. BDNF is expressed by DRG neurons and released in the spinal dorsal horn (Balkowiec and Katz 2000), where it can act on pre- and postsynaptic trkB receptors to regulate plasticity of presynaptic afferent fibers (Matayoshi et al. 2005) and postsynaptic dorsal horn neurons (Kerr et al. 1999; Pezet et al. 2002; Garraway et al. 2003). BDNF expression shows considerable plasticity following peripheral injury (Mannion et al. 1999) and nociceptor-specific knockout of BDNF leads to abrogation of many forms of injury-induced pain plasticity (Zhao et al. 2006). BDNF is also a key factor in LTP. In the hippocampus, BDNF is required for the induction of LTP, and during late LTP, dendritic-expressed BDNF appears to play an autocrine role in the maintenance of late-phase LTP (Fig. 3) (Lu et al. 2008). Likewise, BDNF is capable of inducing LTP in dorsal horn neurons (Zhou et al. 2008) linking BDNF-induced pain plasticity to memory-like mechanisms that may be involved in the maintenance of hyperalgesic priming. Indeed, intrathecal injection of a BDNF scavenging agent, trkB-fc, or systemic injection of a trkB antagonist, ANA-12 (Cazorla et al. 2011), blocks hyperalgesia induced by priming agents and prevents the precipitation of a primed state by subsequent PGE2 injection. Significantly, interruption of BDNF/trkB signaling with either trkB-fc or ANA-12 after the establishment of a primed state leads to a resolution of priming precipitated by PGE2 injection (Melemedjian et al. 2013a). This suggests a key role of BDNF/trkB signaling in the maintenance of a primed state. At spinal synapses, BDNF induces phosphorylation and translation of the two major aPKC isoforms found in the CNS, PKMζ and PKCλ (Fig. 3), suggesting a potential link between BDNF/trkB and aPKCs in the regulation of the maintenance of hyperalgesic priming (Melemedjian et al. 2013a). Because precise targets of ZIP remain unknown, these findings point to BDNF/trkB signaling as a therapeutic target for the reversal of established chronic pain states. Importantly, the precise location of BDNF action and trkB signaling in the maintenance of hyperalgesic priming has not been established. While it is tempting to speculate that an autocrine role for BDNF release from dendrites of dorsal horn neurons in the primed state plays a role in maintaining priming, especially based on the role of this mechanism in the maintenance of late LTP in other CNS structures (Lu et al. 2008), this hypothesis requires further experimental exploration.
4.2 Endogenous Opioids, μ-Opioid Receptor Constitutive Activity, and Hyperalgesic Priming
An intriguing question in the neurobiology of hyperalgesic priming is why does the initial hyperalgesia resolve if a primed state can persist for many weeks to months after the initial injury? This is especially interesting when one considers the strong pharmacological parallels between the maintenance of hyperalgesic priming and the maintenance of late LTP (e.g., aPKCs, AMPA receptor trafficking and BDNF/trkB signaling). In other words, if LTP persists and consolidates into late LTP to maintain priming, why does the initial hyperalgesia resolve? One possibility is that endogenous analgesic mechanisms mask the hyperalgesic state. In such a scenario, the precipitation of hyperalgesia in primed animals would be able to override this endogenous mechanism leading to a very long-lasting hyperalgesic state to a normally subthreshold stimulus. As described above, this is precisely what occurs in hyperalgesic priming models. One candidate for endogenous analgesia overriding hyperalgesia is the endogenous opioid system. This system is robust in the dorsal horn with interneurons capable of releasing peptides that act on μ-opioid receptors (MORs) expressed throughout the dorsal horn (Ribeiro-da-Silva et al. 1992; Ma et al. 1997), including on presynaptic nociceptor nerve endings (Schroeder et al. 1991; Schroeder and McCleskey 1993). Exciting new evidence suggests that this mechanism may be at play in hyperalgesic priming.
Animals exposed to a strong inflammatory stimulus demonstrate hyperalgesia that normally resolves within 10–14 days; however, when these same animals are infused with MOR inverse agonists, the initial hyperalgesia is prolonged for the duration of inverse agonist infusion (Corder et al. 2013). Importantly, even when the initial hyperalgesia is allowed to resolve, infusion of MOR inverse agonists immediately precipitates a reinstatement of hyperalgesia, an effect that is absent in sham animals and an effect that is analogous to precipitation of hyperalgesia in primed animals with a subthreshold peripheral stimulus. What governs this effect? Peripheral inflammation, and presumably other nociceptive stimuli, induces a change in spinal MORs such that they now acquire constitutive activity (signaling through G-proteins in the absence of agonist). This MOR constitutive activity causes a tonic inhibition of pain signaling presumably masking a hyperalgesic state that would otherwise persist following the initial insult. When MOR inverse agonists remove this acquired MOR constitutive activity, a cAMP overshoot occurs (a classical cellular sign of opioid dependence) in a Ca2+-dependent adenylyl cyclase 1 (AC1)-dependent fashion that also involves the engagement of NMDA receptors (Fig. 4) (Corder et al. 2013). This leads to the reinstatement of pain in animals that have a previous history of strong nociceptive input (Fig. 4).
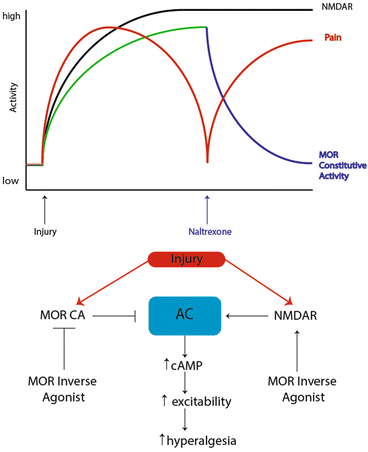
Fig. 4
Signaling pathways regulating endogenous opioid control of hyperalgesic priming. Prior to injury, MORs do not have constitutive activity and nociceptive input to the CNS is absent. However, after injury, nociceptor firing induces activity in CNS circuits leading to release of endogenous opioid peptides in the dorsal horn and the eventual development of MOR constitutive activity resulting in agonist-independent modulation of pain responses. Upon infusion of the MOR inverse agonist naltrexone, AC1 is disinhibited, likely in an NMDA receptor-dependent fashion, allowing for superactivation of cAMP and the reinstatement of hyperalgesia. Adapted from Corder et al. (2013)
These findings have several important implications for understanding central mechanisms governing hyperalgesic priming. First, they provide an elegant solution to the question of why initial hyperalgesia resolves despite the persistence of a primed state. This occurs, at least in part, because tonic opioid signaling, including MOR constitutive activity, masks the presence of mechanisms that would otherwise drive hyperalgesia (Corder et al. 2013). Second, this study provides some possible links to priming and late LTP maintenance that potentially solve questions stated above. Opioid-dependent mechanisms play an important role in governing spinal LTP. While there is evidence that high-dose opioids can stimulate LTP at certain synapses after their abrupt removal (Drdla et al. 2009), there is likewise evidence that MOR activation can resolve even late LTP at spinal synapses (Drdla-Schutting et al. 2012). Therefore, it is formally possible that the initial priming stimulus leads to late LTP consolidation, but this is subsequently resolved by endogenous opioid-mediated mechanisms. A key question then is: does the previous establishment of late LTP at central synapses lead to a drop in threshold for establishment of subsequent LTP? If the mechanisms governing the MOR-dependent reversal of spinal late LTP are constitutively expressed, as appears to be the case (Corder et al. 2013), then this may lead to a tonic reversal of late LTP with underlying mechanisms (e.g., aPKC and BDNF/trkB signaling) still in place. While this idea obviously requires extensive experimental work, it could represent an important mechanism linking changes in peripheral sensitivity to CNS plasticity responsible for the maintenance of priming. Reversing these mechanisms could lead to revolutionary new therapeutics with disease-modifying effects on chronic pain.
4.3 Surgery as a Priming Stimulus and the Effects of Opioids
Opioids have been the first line of therapy for moderate to severe acute pain for decades, if not centuries. However, there is abundant preclinical and some clinical evidence suggesting that opioid administration, designed to alleviate acute pain, paradoxically primes the patient and renders them more susceptible to a transition to chronic pain. Studies using rodents have demonstrated a steady reduction in withdrawal thresholds with intrathecal morphine administration, fentanyl boluses, and repeated systemic morphine (Vanderah et al. 2001; Gardell et al. 2002, 2006) or heroin administration (Mao et al. 1995; Celerier et al. 2000, 2001), generating a sensitized state that is presumably independent of noxious stimulation from the periphery. These preclinical studies highlight the neuroplastic changes induced by opioids and provide another mechanism for the induction of a sensitized and/or primed state in animals.
Related posts:





Full access? Get Clinical Tree


