Fig. 2.1
Framework of genetic background influencing the response to analgesic drugs
The second, more traditional, and better established component of pain related genomics is called pharmacogenomics of pain management and aims to characterize how genetic variations contribute to an individual’s sensitivity and response to a variety of drugs important to pain management practice. Pharmacogenomics is traditionally divided into two parts describing genetic variants influencing pharmacokinetics and pharmacodynamics.
The molecular basis for the observed variability in patient response is defined by different forms of the detected genetic variants. These variants, consisting of the interindividual differences in the DNA sequences, produce the individual phenotypes of the human being. There are many different types of genetic variants (see Fig. 2.2). The most common (more than ten million types known so far) are single nucleotide polymorphisms (or SNP), which represent a point mutation (change of one base) in the DNA fragments. Other allelic mutations include insertion or deletion of a single base (indels), multiple, continuous repeats of 2–4 bases (variable number of tandem repeats or VNTR); repeats of longer DNA fragments (micro– and mini–satellites); copy number variants (CNV, deletion or multiplication of large, >1,000 bases fragments of chromosomes); and finally chromosomal aberrations. The genetic variants may produce alterations in the protein’s function through either changes in the protein expression or its structure.
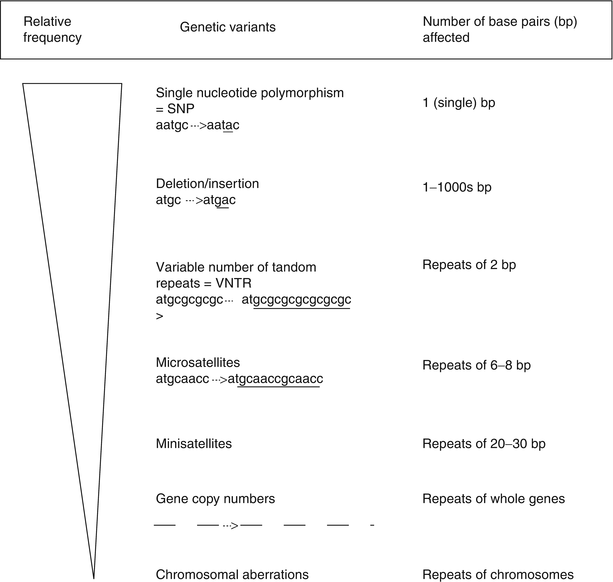
Fig. 2.2
Types of genetic variants taking part in modifying pain phenotype
Functional Genomics of Pain
Pain as a complex trait is expected to have a polygenic nature shaped by the environmental pressures. Identification of specific genetic elements of pain perception promises to be one of the key elements for creating novel and individualized pain treatments. It was demonstrated previously that both rare deleterious genetic variants and common genetic polymorphisms are mediators of human pain perception and clinical pain phenotypes [7, 8]. A higher or lower intensity of pain is very likely to require higher or lower doses of analgesics for efficient pain management. The genetic control of human pain perception and processing is therefore likely to modulate analgesic therapy.
The complete inability to sense pain in an otherwise healthy individual is a very rare phenotype. At present, five types of congenital insensitivity to pain (or HSAN = hereditary sensory and autonomic neuropathy) were identified which are caused by mutations in five different genes [9].
Recently, several new genomic mutations were identified which are described as “channelopathy-associated insensitivity to pain” [10] which are characterized by complete and selective inability to perceive any form of pain. It includes mutations in the alpha-subunit of sodium channel Nav1.7 (SCN9A), causing the loss of function in this specific form of sodium channel [10, 11]. By contrast, mutations in SCN9A that leads to excessive channel activity trigger activation of pain signaling in humans and produce primary erythermalgia (more frequently used term is erythromelalgia), which is characterized by burning pain in response to exposure to mild warmth [12, 13]. Mutations in this gene also produce a rare condition referred to as “paroxysmal extreme pain disorder,” which is characterized by rectal, ocular, and submandibular pain [14].
These syndromes probably have no importance in the everyday clinical pain management as they are very rare, and the affected people probably do not require pain therapy (with exception of erythromelalgia which causes severe pain that is considered a true pain-related emergency). However, defining the molecular causes for hereditary insensitivity to pain may serve as an important source of information to find new targets for analgesic drugs. This assumption was confirmed in the recently published study, in which the authors after investigating 27 common polymorphisms in the SCN9A gene found out that the minor A allele of the SNP rs6746030 was associated with an altered pain threshold and the effect was mediated through C-fiber activation [15]. They concluded that individuals experience differing amounts of pain, per nociceptive stimulus, on the basis of their SCN9A rs6746030 genotype.
Pain in the average population is controlled by fairly frequent genetic variants (allelic frequency > 10 %). Each of them, however, modifies the pain phenotype to only modest degree, and in the majority of cases, the evidence for their involvement in the efficacy of analgesics is either lacking or remains controversial [7, 8]. The involvement of common variants of the opioid receptors, kappa and mu, are discussed below in the part describing pharmacodynamic modifications of activity of opioid analgesics. A variant of third type of opioid receptor, delta, has been associated with lower thermal pain intensity with no association, so far, with the efficacy of opioid analgesics [16].
GTP cyclohydrolase (GCH1), recently implicated in shaping pain responses in humans, regulates production of tetrahydrobiopterin (BH4), an essential factor for the synthesis of dopamine, serotonin, and nitric acid. Tegeder et al. discovered a haplotype associated with reduction of experimental pain in normal volunteers and a favorable outcome with regard to long-term pain reduction that underwent pain (did you mean “a painful surgery”?) surgery [17]. In another study, Tegeder et al. showed that carriers of the particular GCH1 haplotype had higher pain threshold to mechanical and thermal pain following capsaicin sensitization [18]. However, Kim and Dionn and Lazarev et al. failed to replicate significant associations between the same GCH1 genomic variants and pain responses, both in assessment of experimental pain and postoperative pain after dental surgery, as well chronic pancreatic pain [19, 20]. Conversely, the most recent study confirmed again that the five previously identified GCH1 SNPs were profoundly affecting the ratings of pain induced by capsaicin in healthy human volunteers [21]. It was also suggested that the carriers of this particular GCH1 haplotype (which may be responsible for the decreased function of GCH1) display delayed need for pain therapy [2, 22].
Pharmacogenomics of Pain Therapy and Its Usefulness in Clinical Practice
Pharmacogenomics of pain management represents the most familiar area of practical pain genomics. It includes several examples of genomic variations, dramatically changing response to analgesic drugs through either change in their metabolism or receptor targets.
The current list of genetic polymorphisms which may affect the action of analgesic drugs is quite long and appears to be growing rapidly. The best known mechanisms involved in the altered effects of analgesics involve polymorphic changes in its metabolism. In this respect, three major mechanisms have been identified, involving genetic variations in the metabolic activation of the analgesics administered as an inactive or less active prodrug, variations in the metabolic degradation of the active components, and variations in its transmembrane transport.
Genetic Variations in the Prodrug Activation
The better known example involves polymorphisms in genes of the liver isoforms of the cytochrome P450 system (CYP) [23]. In particular, the most well-characterized CYP2D6 polymorphism is responsible for the considerable variation in the metabolism (and clinical responses) of drugs from many therapeutic areas, including several analgesics (Fig. 2.3) [24–26]. More than 100 CYP2D6 alleles have been identified, ranging from nonsynonymous mutations to SNPs that either alter RNA splicing or produce deletions of the entire gene [27]. Of these, *3, *4, and *8 are nonfunctional, *9, *10, and *41 have reduced function, and *1,*2, *35, and *41 can be duplicated, resulting in greatly increased expression of functional CYP2D6. There are also interethnic differences in the frequencies of these variant alleles. Allele combinations determine phenotype: two nonfunctional = poor metabolizer (PM); at least one reduced functional = intermediate metabolizer (IM); at least one functional = extensive metabolizers (EM); and multiple copies of a functional and/or allele with promoter mutation = ultrarapid metabolizer (UM). The most recent update of CYP2D6 nomenclature and terminology could be found on home Web page of the Human Cytochrome P450 (CYP) Allele Nomenclature at http://www.cypalleles.ki.se/.
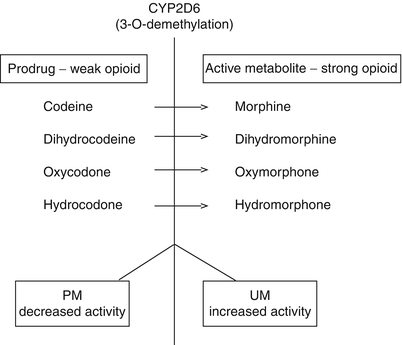
Fig. 2.3
Opioid analgesics influenced by polymorphic CYP2D6 metabolism (3-O-demethylation)
Codeine and other weak opioids are extensively metabolized by polymorphic CYP2D6 which regulates its O-demethylation to more potent metabolites (e.g., after a single oral dose of 30 mg codeine, 6 % is eventually transformed to morphine). The clinical analgesic effect of codeine is mainly attributed to its conversion to morphine, which has a 200 times higher affinity and 50 times higher intrinsic activity at MOR than codeine itself [28]. Since CYP2D6 is genetically highly polymorphic, the effects of codeine are under pharmacogenetic control.
Genetically, altered effects of codeine may occur in subjects with either decreased, absent, or highly increased CYP2D6 activity when compared with the population average [29, 30]. Decreased or absent CYP2D6 activity in PMs causes production of only very low or absent amount of morphine after codeine administration. The ultrafast metabolizers (UM) produce on the other hand excessive amount of morphine after typical dose of codeine. Roughly, one out of seven Caucasians is at risk of either failure or toxicity of codeine therapy due to extremely low or high morphine formation, respectively. Recent case reports of codeine fatalities highlighted that the use of this weak opioid, particularly in young children, is associated with a substantial risk in those subjects displaying UM genotype [31–37]. The polymorphic variants in the CYP2D6 system are responsible for some but not all variability observed after codeine administration. The other causes for the observed high variability in codeine efficacy include both polymorphisms in other genes involved in opioid expression or trafficking, as well as nongenetic factors. In addition to differences in codeine metabolism between EM and PM, the differences between EM of various ethnicities have also been highlighted. The Chinese EM reported having a lower rate of codeine O-demethylation when compared with the Caucasian EM, because of the much higher frequency (50 %) of the *10 (reduced function) allele in the Chinese [27].
Other popular analgesic drug which depends on activation by the CYP2D6 includes tramadol. Tramadol is a mu-opioid receptor (MOR) agonist but has a lower affinity at MORs than its active metabolite O-desmethyltramadol. Tramadol itself has weak analgesic activity which becomes evident when CYP2D6 is blocked and also acts through non-opioid-dependent mechanisms which involve serotonin- and noradrenaline-mediated pain inhibition originating in brain stem. The analgesic activity of tramadol is strongly modulated by CYP2D6 activity. The analgesic activity on experimental pain is reduced in CYP2D6 PMs, a finding later confirmed in pain patients [38–42]. It is interesting to note that the pharmacokinetics of tramadol (which is administered as racemic substance containing equal amount of (−) and (+) optical isomers producing analgesia by a synergistic action of its two enantiomers and their metabolites) is enantioselective in CYP2D6 poor and extensive metabolizers, meaning that the production of either optical isomer differs depending on the metabolic status [41, 43, 44]. The clinical significance of this finding for pain management remains to be further explored.
As far as other CYP2D6 substrates with active analgesic metabolites are concerned (see Fig. 2.3), the evidence for variation in its analgesic with altered CYP2D6 function is less evident when compared with codeine or tramadol. These examples are either negative, such as for dihydrocodeine [45, 46], or explained at a nongenetic level, such as for oxycodone. In other cases, the evidence is based only on animal studies, such as hydrocodone, or restricted to single case reports for inadequate activity of oxycodone [3].
Tilidine (an opioid analgesic) is activated to active metabolite nortilidine, and parecoxib (an NSAID) is activated into valdecoxib by CYP3A system. This enzyme is phenotypically highly variable, but only a minor part of this variability can be attributed to genetics [2]. Individuals with at least one CYP3A5*1 allele copy produce fully active active copy of CYP3A5 enzyme; however, the majority of Caucasians have no active CYP3A5 due to a premature stop codon.
Genetic Variations in the Elimination of Analgesic Drugs
Many of the opioids contain hydroxyl group at position 6, and the potent opioids have a hydroxyl at position 3 of the 4,5-methoxymorphinan structure. The glucuronidation of morphine, codeine, buprenorphine, dihydrocodeine, dihydromorphine, hydromorphone, dihydromorphine, oxymorphone, as well as opioid receptor antagonists (naloxone and naltrexone) is mainly mediated by the uridine diphosphate (UDP) glucuronyltransferase (UGT)2B7 [3]. Similar to CYP genes, the UGT2B7 gene is also polymorphic, although less than 20 allelic variants have been identified. The main proportion of morphine is metabolized to morphine-6-glucuronide, M6G (approximately 70 %), and to lesser degree to morphine-3-glucuronide (M3G). Both metabolites are active, with effects opposite to each other, consisting in excitation and anti-analgesia for M3G and in typical opioid agonist effects for M6G. Despite the role of UGT2B7 in the formation of M6G and M3G, the clinical effect of the UGT2B7*2 (268Y) variant has only produced conflicting results so far. Different variants in the 5′ untranslated region of UGT2B7 are associated with reduced M6G/morphine ratios in patients. In addition, it was reported that UGT2B7 *2/*2 genotypes and CYP2D6 UM phenotypes were associated with severe neonatal toxicity after breast-feeding and oral ingestion of opioids. The above preliminary data indicate that the consequences of UGT variants were so far restricted to alterations of plasma concentrations, while none of the UGT variants alone have been associated with the altered efficacy of opioid analgesics [3, 4].
The increased enzyme activity associated with the CYP3A5*1 allele may cause accelerated elimination of CYP3A substrates, such as alfentanil, fentanyl, or sufentanil. However, positive associations of CYP3A polymorphisms with analgesic actions have not been reported so far. The CYP3A5 genotype did not affect the systemic or apparent oral clearance as well as the pharmacodynamics of alfentanil and levomethadone [47, 48].
In addition to CYP2D6 and CYP2A5, there is also clinical evidence about the involvement of other CYP systems in the metabolism of frequently used nonsteroid anti-inflammatory drugs (NSAIDs). Human CYP2C9 metabolizes numerous drugs (e.g., warfarin, oral sulfonylurea hypoglycemics, antiepileptics, and others) [49]. In addition, CYP2C9 polymorphism might play a significant role in the analgesic efficacy and toxicity of traditional NSAIDs, for example, diclofenac, ibuprofen, naproxen, tenoxicam, and piroxicam, as well as selective COX-2 inhibitors such as celecoxib and valdecoxib [50]. More than 33 variants and a series of subvariants have been identified for CYP2C9 to date. The two missense mutations, CYP2C9*2 (rs1799853) and CYPC9*3 (rs1057910), yield enzymes with decreased activity [51]. These alleles are mainly present in Caucasians, while their frequency is lower in African and Asian subjects. More than twofold reduced clearance after oral intake of celecoxib was observed in homozygous carriers of CYP2C9*3 compared with carriers of the wild-type genotype CYP2C9*1/*1 [52]. Similarly, ibuprofen-mediated inhibition of COX-1 and COX-2 is significantly decreased (by 50 %) in carriers of two CYP2C9*3 alleles [53]. Further investigations demonstrating the relevance of the CYP2C9*3 allele for naproxen, tenoxicam, piroxicam, and lornoxicam pharmacokinetics have been also published [4, 54]. Although CYP2C9 is the major determinant of clearance, it is necessary to also consider CYP2C8 genotype, as it contributes to some smaller extent in NSAIDs metabolism. In the study performed in healthy volunteers, it was demonstrated that metabolism of diclofenac was significantly slower in individuals carrying CYP2C8*3 (rs10509681) or CYP2C8*4 (rs1058930) allele than in those homozygous for the wild-type allele [55].
Whereas numerous clinical trials have demonstrated the impact of CYP2C9*3 on therapy with Coumadin, less information is available on the CYP2C9 genotype-related efficacy of NSAIDs in the pain management. Some publications focus, however, on the incidence and severity of adverse effects (e.g., gastrointestinal (GI) bleeding, effects on coagulation). It was that the combined presence of CYP2C8*3 and CYP2C9*2 was a relevant determinant in the risk of developing GI bleeding in patients receiving NSAIDs metabolized by CYP2C8/9 [56]. Similar results were also presented by Agundez et al. [57]. However, to date, the study results from other authors are conflicting, with several other trials reporting no association [58, 59]. More studies are clearly necessary to confirm the relevance of CYP2C8/9 genotype with increased incidence of GI bleeding.
Another typical adverse effect is the influence of classical NSAIDs on coagulation. The risk of altered coagulation was substantially increased in patients with either CYP2C9*3 and CYP2C9*3 (mentioned twice) genotypes taking Coumadin together with NSAIDs which are known CYP2C9 substrates [60].
Genetic Variations in the Transmembrane Transport of Analgesics
P-glycoprotein (P-gp) coded by the ATP-binding cassette subfamily (ABCB1)/multidrug resistance (MDR1) gene is mainly located in organs with excretory functions (e.g., liver, kidneys). It is also expressed at the blood-brain barrier where it forms an outward transporter. Therefore, functional impairment of P-gp-mediated drug transport may be expected to result in increased bioavailability of orally administered drugs, reduced renal clearance, or an increased brain concentration of its substrates. Some opioids are P-gp substrates. The ABCB1 3435 C > T variant (rs1045642) is associated with decreased dosage requirements in opioids that are P-gp substrates, as assessed in outpatients. Moreover, a diplotype consisting of three polymorphic positions in the ABCB1 gene (1236TT-rs1128503, 2677TT-rs2032582, and 3435TT) is associated with increased susceptibility to respiratory depression caused by fentanyl in Korean patients [61]. The results suggest that analysis of ABCB1 polymorphisms may have clinical relevance in the prevention of respiratory suppression by intravenous fentanyl or to anticipate its clinical effects. With the OPRM1 118 A > G variant (see below), the ABCB1 3435 C > T predicted the response to morphine in cancer patients with a sensitivity close to 100 % and a specificity of more than 70 % [62]. Trials in patients suffering from chronic and cancer pain had shown decreased opioid consumption in carriers of the 3435T allele [63, 64]. Finally, methadone analgesia may be subject to P-gp pharmacogenetic modulation. The pupillary effects of orally administered methadone are increased following the pharmacological blockade of P-gp by quinidine, and the methadone dosing for heroin substitution can be decreased in carriers of ABCB1 variants associated with decreased transporter expression, for example, ABCB1 2435 C > T and others [65, 66].
Pharmacodynamics of Pain Therapy
The alterations in effects of analgesics may also result from pharmacodynamic interferences, consisting of altered receptor binding, activation or signaling mechanisms, or of altered expression of the drug’s target, such as opioid receptors or cyclooxygenases. Genetic factors have been found to act via any of these mechanisms.
Opioid Receptors
The mu-opioid receptor (MOR) is part of the family of several types of opioid receptors which are 7-transmembrane domain, G-protein-coupled receptors (GPCR), and inhibit cellular activity. MOR is clinically most relevant target of opioid analgesics. The OPRM1 gene coding for MOR in humans is highly polymorphic, with excess of 1,800 SNPs listed in the current edition (2010) of the NCBI SNP database (http://www.ncbi.nlm.nih.gov/snp). Coding mutations affecting the third intracellular loop of MOR (e.g., 779 G > A, 794 G > A, 802 T > C) result in reduced G-protein coupling, receptor signaling, and desensitization, leading to an expectation that opioids should be almost ineffective in patients carrying those polymorphisms. However, these polymorphisms are extremely rare (<0.1 % population) and are therefore restricted to very rare single cases.
Evidence for a function of OPRM1 variants with allelic frequencies >5 % is sparse, except for the 118 A > G polymorphism (rs1799971). This SNP causes an amino acid exchange of the aspartate with an asparagine at position 40 of extracellular part of MOR, deleting one of a putative glycosylation sites. This change can cause altered expression of MOR or its signaling [67–69]. The OPRM1 118 A > G polymorphism has an allele frequency of 8–17 % in Caucasians and considerably higher in Asians, with a frequency of 47 % reported from Japan. It is also worth noting that the frequency of homozygotes for the GG allele is by much higher in Asian population with only very rare (<1 %) occurrence in Caucasian population [70]. The data obtained so far with the OPRM1 118 A > G polymorphism have been controversial [71]. The molecular changes associated with SNP 118 A > G translate to a variety of clinical effects (predominantly decrease) of many opioids in experimental settings and clinical studies [72–81]. The consequences of the SNP 118 A > G have consistently been related to a decrease in opioid potency for pupil constriction (e.g., for morphine, M6G, methadone). For analgesia, the SNP decreases the concentration-dependent effects of alfentanil on experimental pain. Specifically, the variant decreases the effect of opioids on pain-related activation mainly in those regions of the brain that are processing the sensory dimension of pain including the primary and secondary somatosensory cortex and posterior insular cortex [82]. In clinical settings, greater postoperative requirements of alfentanil and morphine have been reported for carriers of the variant, and higher concentrations of alfentanil of M6G were needed to produce analgesia in experimental pain models [2, 48, 83–87]. It should be noted that other studies described only moderate to no significant effects of the OPRM1 118 A > G polymorphism on opioid requirements or pain relief. Several studies did not demonstrate any association between OPRM1 variant and analgesic needs [88–92]. Contradictory results were reported by Landau et al. who investigated the influence of OPRM1 118 A > G polymorphism on the analgesic effectiveness of fentanyl in females after its intrathecal administration during labor and delivery. The analgesic requirements in this study were increased in homozygous carriers of AA allele, the opposite effect compared with most other studies [93]. In the chronic pain patients, it was reported that in the high-quartile opioid utilization group, the homozygous carriers of the minor allele required significantly higher opioid doses than the carriers of the minor allele [91]. In another studies, GG homozygote patients were characterized by higher morphine consumption than carriers of the major AA allele [76, 94]. In summary, an influence of OPRM1 genetic variants on opioid requirements and degree of pain relief under opioid medication has been demonstrated in some studies; however, this could not be replicated in all subsequent investigations. Patients stratification; a low number of patients with the GG genotype (in particular in studies performed in Caucasian populations); presence of multiple, uncontrolled co-variables influencing the phenotype; and a clinically questionable reduction in opioid consumption are some major concerns. The requirements of high opioid doses may in part reflect an addiction component or a higher/faster rate of tolerance development in certain pain patients. It was reported that OPRM1 A118G polymorphism is a major determinant of striatal dopamine responses to alcohol. Social drinkers recruited based on OPRM1 genotype were challenged in separate sessions with alcohol and placebo under pharmacokinetically controlled conditions and examined for striatal dopamine release using positron emission tomography and [(11)C]-raclopride displacement. A striatal dopamine response to alcohol was restricted to carriers of the minor 118G allele. Based on the results of this study, it was concluded that OPRM1 A118G variation is a genetic determinant of dopamine responses to alcohol, a mechanism by which it likely modulates alcohol reward [95].
In addition, the most recent study seems to suggest that some of the effect of SNP A > G could be explained by the linkage disequilibrium with other functional SNPs located in the OPRM1 region [96]. For example, SNP rs563649 is located within a structurally conserved internal ribosome entry site in the 5′-UTR of a novel exon 13-containing OPRM1 isoforms (MOR-1K) and affects both mRNA levels and translation efficiency of these variants. Furthermore, rs563649 exhibits very strong linkage disequilibrium throughout the entire OPRM1 gene locus and thus affects the functional contribution of the corresponding haplotype that includes other functional OPRM1 SNPs. These results might provide evidence for an essential role for MOR-1K isoforms in nociceptive signaling and suggest that genetic variations in alternative OPRM1 isoforms may contribute to individual differences in opiate responses.
Catechol-O-Methyltransferase (COMT)
CMOT degrades catecholamine neurotransmitters such as norepinephrine, epinephrine, and dopamine. Increased dopamine concentrations suppress the production of endogenous opioid peptides. Opioid receptor expression is in turn upregulated, which has been observed with the Val158Met variant of COMT, coded by the COMT 772 G > A (rs4680) SNP in human postmortem brain tissue and in vivo by assessing radiolabeled 11C-carfentanil MOR binding [97, 98]. This variant leads to a low-function COMT enzyme that fails to degrade dopamine, which may cause a depletion of enkephalin. Patients with cancer carrying the Val158Met variant needed less morphine for pain relief than patients not carrying this variant. Finally, the variants exerts its opioid enforcing effects also in cross relation with the OPRM1 118 A > G variant [94, 97–101]. During the past decade, several new polymorphisms were identified in the COMT gene which contains at least five functional polymorphisms that impact its biological activity and associated phenotypes (including pain). The potentially complex interactions of functional variations in COMT imply that the overall functional state of the gene might not be easily deduced from genotype information alone, which presumably explains the inconsistency in the results from association studies that focus on the V158Met polymorphism [102, 103].
Melanocortin 1 Receptor (MC1R)
Nonfunctional variants of the MC1R which produces bright red hair and fair skin phenotype were associated with an increased analgesic response to kappa opioid receptors (KOR)-mediated opioid analgesia. Red-headed women required less of the KOR agonist drug – pentazocine – to reach a specific level of analgesia compared with all other groups [104, 105]. This study presented the first strong evidence for a gene-by-sex interaction in the area of pain genetics, because the authors also showed that red-headed men did not experience enhanced KOR analgesia.
Cyclooxygenases (COX)
Polymorphisms in the prostaglandin endoperoxidase synthase 2 gene (PTGS2) coding for COX-2 may modulate the development of inflammation and its response to treatment with inhibitors of COXs, especially those specific for COX-2 [106]. This has been proposed for the PTGS2-765 G > C SNP (rs20417), which was reported to be associated with more than a twofold decrease in COX-2 expression [107]. By altering a putative Sp1 binding site in the promoter region of PTGS2, this gene variant was found to decrease the promoter activity by 30 % [108]. However, the controversial results were reported so far in clinical studies with this polymorphisms and different COX-2 inhibitors. The inhibitory effect of celecoxib on COX-2 was not associated with the presence of this variant in volunteers [109]; conversely, significantly decreased analgesic effects of rofecoxib were observed in the homozygous carriers of this variant [110].
Related posts:
 The Role of Antidepressants in the Treatment of Chronic Pain
Anticonvulsant Medications for Treatment of Neuropathic and “Functional” Pain
Polypharmacy and Drug Interaction
The Role of Antidepressants in the Treatment of Chronic Pain
Anticonvulsant Medications for Treatment of Neuropathic and “Functional” Pain
Polypharmacy and Drug Interaction
 Opioid Adverse Effects and Opioid-Induced Hypogonadism
Opioid Adverse Effects and Opioid-Induced Hypogonadism
 Clinical Use of Opioids
Clinical Use of Opioids
 Acute Management of the Opioid-Dependent Patient
Acute Management of the Opioid-Dependent Patient

Full access? Get Clinical Tree


