Permissive Hypercapnia: Introduction
Carbon dioxide (CO2) is the “waste product” of aerobic respiration. In health, arterial carbon dioxide tension (PaCO2) is tightly regulated, with minute ventilation potently enhanced in response to small elevations in CO2 tension. In critical illness its role is becoming better understood; indeed, in survivors of cardiac arrest, elevated CO2 is associated with a higher incidence of reported near-death experiences.1 Although usually well tolerated, hypercapnia traditionally has been considered to be an adverse event. In fact, the extent and severity of acidosis are predictive of adverse outcome in diverse clinical contexts, including cardiac arrest,2,3 sepsis,4–6 and in neonatal practice.7 Traditional approaches to CO2 management in the operating room and for patients with acute respiratory failure generally focused on the deleterious effects of hypercapnia, traditionally targeting therefore normocapnia or even hypocapnia.
This approach, however, has been increasingly questioned. The potential for high tidal volumes to injure the lung directly, a phenomenon termed ventilator-induced lung injury, is clear from experimental8,9 and clinical10–14 studies. Current “protective” ventilator strategies mandate lower tidal volumes (VT), and generally necessitate hypoventilation and tolerance of hypercapnia. This “permissive hypercapnia” has been accepted progressively in critical care for adult, pediatric, and neonatal patients requiring mechanical ventilation.
Rationale
The potential for mechanical ventilation to contribute to lung and systemic organ injury and to worsen outcome in patients with the acute respiratory distress syndrome (ARDS) is clear. The use of high VT may cause injury via several mechanisms.8,9 Increased mechanical stress may activate the cellular and humoral immune response directly in the lung.8,15–17 Intrapulmonary mediators and pathogens, such as prostaglandins,18 cytokines,19 endotoxins,20 and bacteria,21 have been demonstrated to access the systemic circulation following high-stretch mechanical ventilation. The demonstration that mechanical ventilation may cause systemic organ dysfunction in animal models could explain in part the high rate of multiorgan failure in ARDS.22 In current practice, hypercapnia is tolerated as the lesser of two evils so as to realize the benefits of lower tidal volumes.23,24
Basic Principles
Conventionally, the protective effect of ventilator strategies incorporating permissive hypercapnia is solely secondary to lower tidal volume, with hypercapnia being permitted so as to achieve this goal. Protective ventilator strategies that involve hypoventilation cause both limitation of lung stretch and elevation of arterial PCO2; thus, low tidal volume is distinct from elevated PCO2 and, by manipulation of respiratory variables (frequency, VT, dead space, and inspired CO2), can to some extent be controlled separately.
If hypercapnia were proven to have independent benefit, then deliberately elevating PaCO2, termed therapeutic hypercapnia, might provide an additional advantage over reducing VT alone. Conversely, in patients managed with conventional permissive hypercapnia, adverse effects of elevated PaCO2 may be concealed by the benefits of lessened lung stretch. These issues are underscored by the fact that hypercapnia has adverse effects in some clinical settings, such as critically elevated intracranial pressure or pulmonary vascular resistance. Because patient outcome from critical illness may be related to systemic rather than lung injury, the clinician also must consider the effects on the brain and cardiovascular systems.
These issues have led several investigators to study the direct effects of induced hypercapnia per se in models of lung and systemic organ injury. These studies raise the potential that hypercapnia may exert active roles—beneficial or adverse—in the pathogenesis of inflammation and tissue injury. Thus, therapeutic hypercapnia may someday become a testable clinical approach to critically ill patients.25
This is reported most commonly in the context of errors in mechanical ventilation, such as circuit disconnects (or malconnections that permit rebreathing of exhaled gases).26 A second cause is hypoventilation secondary to drug-induced respiratory depression,27 severe respiratory failure (e.g., status asthmaticus),28 or massive aspiration (e.g., grain inhalation).29
The determinants of hypercapnia can be described as the balance of CO2 production versus elimination.
Inspired CO2 (FIO2) usually is negligible. Increased CO2 production is a potential contributor to hypercapnia, particularly in hypermetabolic disease states such as hyperpyrexia. Therefore, for practical purposes PaCO2, reflects the rate of elimination of CO2; there are no common “physiologic” causes of hypercapnia. Hypercapnia is seen most commonly in the context of acute or chronic respiratory failure associated with reduced minute ventilation or increased dead space.
The effects of hypercapnia on systemic oxygenation are discussed below. It is often forgotten that decreasing VT can reduce oxygenation of pulmonary venous blood. This has two contributing elements: the composition of the alveolar gas and the increase in intrapulmonary shunt. Decreased VT increases propensity to atelectasis, which increases intrapulmonary shunt.30 In addition, when VT is decreased, there is a proportionate decrease in alveolar ventilation (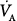 ), resulting in an elevated alveolar CO2 tension (PaCO2). As pointed out by Bidani et al,31 the alveolar gas equation
), resulting in an elevated alveolar CO2 tension (PaCO2). As pointed out by Bidani et al,31 the alveolar gas equation
where PAO2 is the alveolar oxygen tension, PB is the barometric pressure, and R is the respiratory quotient, can be combined with the following relationship:
where 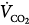 is CO2 production, to express alveolar O2 as a function of FIO2 and
is CO2 production, to express alveolar O2 as a function of FIO2 and 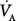 :
:
Thus, with the exception of very low levels of minute ventilation, increases in  have minor impact on alveolar—and consequently pulmonary venous—oxygenation. In contrast, modest increases in FIO2 can have a far more significant impact on oxygenation and can compensate easily for reduced VA (Fig. 14-1).31
have minor impact on alveolar—and consequently pulmonary venous—oxygenation. In contrast, modest increases in FIO2 can have a far more significant impact on oxygenation and can compensate easily for reduced VA (Fig. 14-1).31
Figure 14-1
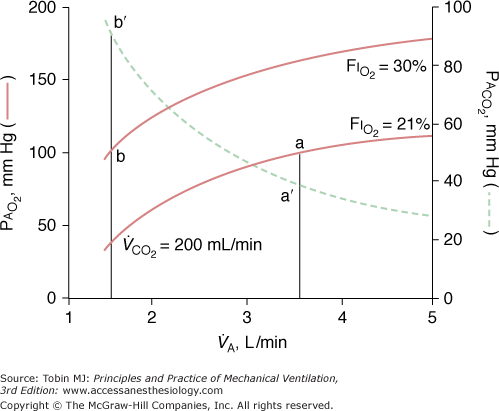
Relationship of alveolar oxygen tension (PAO2) and alveolar ventilation (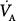 ) at FIO2 (0.21 and 0.30), calculated assuming a constant carbon dioxide production (
) at FIO2 (0.21 and 0.30), calculated assuming a constant carbon dioxide production (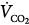 of 200 mL/min). At either FIO2, decreasing
of 200 mL/min). At either FIO2, decreasing 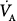 to low values significantly reduces PAO2, especially with very low levels of
to low values significantly reduces PAO2, especially with very low levels of  . A small increase in FIO2 (21% to 30%) easily compensates for the fall in PAO2 resulting from hypoventilation. Solid line, relationship between PAO2 and
. A small increase in FIO2 (21% to 30%) easily compensates for the fall in PAO2 resulting from hypoventilation. Solid line, relationship between PAO2 and  ; dotted line, relationship between PaCO2 and
; dotted line, relationship between PaCO2 and  . (Modified, from Bidani A, Tzouanakis AE, Cardenas VJ Jr, Zwischenberger JB. Permissive hypercapnia in acute respiratory failure. JAMA. 1994;272:957–962.)
. (Modified, from Bidani A, Tzouanakis AE, Cardenas VJ Jr, Zwischenberger JB. Permissive hypercapnia in acute respiratory failure. JAMA. 1994;272:957–962.)
Physiologic Effects
The physiologic effects of hypercapnia are complex and incompletely understood, with direct effects often counterbalanced by indirect effects. To understand hypercaponia in critically ill patients, the effects of pH and PaCO2 need to be considered together.
Hypercapnia generally results in acidosis (greater H+ concentration) via its spontaneous and carbonic anhydrase–catalyzed combination with water to form carbonic acid. The protons thus generated can react with titratable groups in certain amino acids, resulting in structural changes in many proteins and enzymes in cell membranes and cellular aqueous environments.32 Because acidosis suppresses most cellular functions, the body uses a number of strategies to defend its intracellular and extracellular pH within remarkably narrow limits.25 The intracellular acidosis produced by hypercapnia may be corrected within a few hours, as opposed to 1 to 2 days for renal compensation.33 This buffering occurs via active cell-membrane ion transporters that extrude protons and exchange them for extracellular sodium.
During hypercapnia, CO2 per se may react directly with some free amine groups in proteins to form carbamate residues.34–36 This binding of CO2 also modifies protein structure and function and may explain some of the differences in the observed effects of CO2 and H+ when both lead to equal changes in pH. A good example is the Bohr effect, where increased PaCO2 results in a rightward shift of the hemoglobin (Hb)-O2 dissociation curve, reflecting a lowered affinity of hemoglobin for O2.
In contrast to the systemic circulation, hypercapnic acidosis produces vasoconstriction and increased resistance in the pulmonary circulation;37 such effects are exacerbated in the setting of preexisting pulmonary hypertension.37 Hypercapnic vasoconstriction generally is weaker than hypoxic pulmonary vasoconstriction; its more important effect may be in augmenting hypoxic vasoconstriction.32 Little is understood about how CO2 acts on pulmonary vascular smooth muscle.
Acute respiratory acidosis can alter shunt via autonomic or direct effects on pulmonary vasculature and on the airways. Acidosis enhances hypoxic pulmonary vasoconstriction and therefore usually reduces shunt and increases partial pressure of arterial oxygen (PaO2), whereas alkalosis has the opposite effect.38 CO2 administration improves matching of ventilation and perfusion and increases arterial oxygenation by this mechanism in both health39–41 and disease.42 A dose–response relationship exists wherein increased FICO2 results in progressive augmentation of PaO2.40,43 In fact, administration of CO2 during late inspiration—limiting its exposure to the conducting airways—results in most of the beneficial pulmonary effects (i.e., 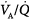 matching and oxygenation) and less systemic acidosis (Fig. 14-2).44 Permissive hypercapnia in patients with ARDS appears to increase shunt secondary to a reduction in VT and airway closure rather than from elevated CO2 levels.45
matching and oxygenation) and less systemic acidosis (Fig. 14-2).44 Permissive hypercapnia in patients with ARDS appears to increase shunt secondary to a reduction in VT and airway closure rather than from elevated CO2 levels.45
Figure 14-2

Effects of room air and inspired CO2 on the distribution of ventilation (A) and of perfusion (B). The lowest values of 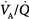 correspond to regions of intrapulmonary shunt and the highest to regions of alveolar dead space. Addition of CO2 to inspired gas (throughout the respiratory cycle, *; restricted to late inspiration, □) resulted in more homogeneous ventilation and perfusion compared to no added CO2 (room air ♦).
correspond to regions of intrapulmonary shunt and the highest to regions of alveolar dead space. Addition of CO2 to inspired gas (throughout the respiratory cycle, *; restricted to late inspiration, □) resulted in more homogeneous ventilation and perfusion compared to no added CO2 (room air ♦). 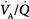 , ratio of ventilation to perfusion. (Reproduced, with permission, from Brogan TV, Robertson HT, Lamm WJ, Souders JE, Swenson ER. Carbon dioxide added late in inspiration reduces ventilation–perfusion heterogeneity without causing respiratory acidosis. J Appl Physiol. 2004;96:1894–1898.)
, ratio of ventilation to perfusion. (Reproduced, with permission, from Brogan TV, Robertson HT, Lamm WJ, Souders JE, Swenson ER. Carbon dioxide added late in inspiration reduces ventilation–perfusion heterogeneity without causing respiratory acidosis. J Appl Physiol. 2004;96:1894–1898.)
Hypercapnia has been reported to either increase46 or decrease47 airway resistance. These effects may be explained the direct dilation of small airways and the indirect (i.e., vagally mediated) large airway constriction.32 These opposing but balanced actions may produce little net alteration in airway resistance.48
Parenchymal lung compliance increases in response to hypercapnic acidosis. This may be secondary to increased surfactant secretion or more effective surface tension–lowering properties under acidic conditions.49
In health, CO2 alters lung compliance as well as pulmonary vascular and airway tone.32 The combined effect of small airways constriction and decreased compliance explains the phenomenon of hypocapnic bronchoconstriction and pneumoconstriction that occurs following acute regional pulmonary artery occlusions.50,51 These effects either may alter regional ventilation to keep pace with a primary change in perfusion or may alter regional perfusion to match a primary change in ventilation.32
Hypercapnic acidosis causes cerebral vasodilation. The increase in cerebral blood flow and blood volume must be considered carefully in any patient at risk for raised intracranial pressure.25 The mechanism of cerebral vasodilation depends on the arterial bed and type of artery. Nakahata et al52 demonstrated that hypercapnic acidosis induced cerebral precapillary arteriolar vasodilation, which depended on acidosis rather than CO2. They demonstrated that the adenosine triphosphate (ATP)-sensitive potassium channel plays a major role. Others have suggested roles for both ATP-sensitive and calcium-activated potassium channels53 and the neuronal isoform of nitric oxide synthase.54
Hypercapnia is a potent regulator of ventilation. Mild hypercapnia (increase in end-tidal pressure of carbon dioxide [PCO2] of 8 mm Hg) in healthy volunteers resulted in a compensatory metabolic alkalosis over 24 to 48 hours that was maintained over the course of exposure.55 Although there was a modest increase in ventilatory chemosensitivity to acute hypoxia, no change occurred in response to acute elevations in CO2.
Hare et al40 demonstrated that hypercapnic acidosis increases cerebral tissue partial pressure of oxygen (PO2) through augmentation PaO2 of and increased cerebral blood flow (Fig. 14-3), although, studies in patients suggest that in the presence of sepsis, hypercapnia may impair cerebral autoregulation.56 The faster recovery times associated with administered CO2 in animals57 and patients58 following general anesthesia may reflect increased perfusion and anesthetic washout.
Figure 14-3

Increases in inspired CO2 concentration produce progressive increases in brain tissue O2 tension and cerebral perfusion. (Reproduced, with permission, from Hare GM, Kavanagh BP, Mazer CD, et al. Hypercapnia increases cerebral tissue oxygen tension in anesthetized rats. Can J Anaesth. 2003;50:1061–1068.)
Recent studies highlight the potential for hypercapnia to exert deleterious neuromuscular effects. Rat diaphragms exposed to prolonged hypercapnia (7.5% CO2 for 6 weeks) undergo significant changes,59 including depressed diaphragmatic tension and time to contraction and relaxation, and altered diaphragmatic composition (i.e., increased slow-twitch and decreased fast-twitch fibers). In fact, even short-term exposure (7% inspired CO2) may impair neuromuscular function transiently through effects on afferent transmission or synaptic integrity in healthy volunteers.60 Relatively brief exposures to hypercapnia may also cause reversible impairment of diaphragmatic muscle contractility.61
Hypercapnic acidosis directly reduces the contractility of cardiac and vascular smooth muscle.32 This is counterbalanced by the hypercapnic-mediated sympathoadrenal effects causing increased preload, increased heart rate, and decreased afterload, which lead to a net increase in cardiac output.32 In intact animals and human subjects, myocardial contractility and cardiac output increase during hypercapnia because of increased sympathetic activity.62
Hypercapnia results in a complex interaction of altered cardiac output, hypoxic pulmonary vasoconstriction, and intrapulmonary shunt, with a net increase in PaO2. Because hypercapnia generally elevates cardiac output, global O2 delivery is increased.63 Regional (including mesenteric) blood flow also is increased,64 thereby increasing organ oxygen delivery. Because hypercapnia and acidosis shift the Hb-O2 dissociation curve rightward and may cause an elevation in hematocrit,65 tissue oxygen delivery is further facilitated. Acidosis may reduce cellular respiration and oxygen consumption,66 which further correct supply–demand imbalance, in addition to enhancing O2 delivery. Acidosis may protect against ongoing tissue production of further organic acids by a negative-feedback loop, providing a mechanism of cellular metabolic shutdown at times of nutrient shortage (e.g., ischemia).67 In addition, hypercapnic acidosis increases PO2 in subcutaneous tissues and in the intestinal wall.68
Hypercapnia may exert complex effects on development,69 such as inhibition of embryonic morphogenesis, as well as egg laying and hatching in the Drosophila.70 It induces aberrant motility and slows development, decreases fertility, and increases life span in Caenorhabditis elegans.71 Tissue effects in neonates may differ from those in adults as exposure to hypercapnia for 2 weeks reduced expression of several matrix proteins in infant, but not in adult, lungs,72 and sustained exposure in the neonate may cause microvascular injury and impair brain growth via induction of nitrative stress.73 In the neonatal lung, hypercapnia may increase alveolar budding, but may also increase central nervous system apoptosis.74
Cellular and Molecular Effects of Hypercapnia
A clear understanding of cellular and biochemical mechanisms underlying the effects of hypercapnia is essential for successful translation of laboratory findings to the bedside, as well as for prediction of potential side effects.
Hypercapnia and acidosis modulate diverse components of the host immune response, particularly cytokine and chemokine signaling, as well as neutrophil and macrophage function.
Hypercapnia interferes with coordination of the immune response by modulating signaling among immune effector cells. Hypercapnic acidosis inhibits neutrophil release of interleukin (IL)-8 following endotoxin stimulation75 (Fig. 14-4), reduces release of tumor necrosis factor-α (TNF-α) and IL-1 from stimulated macrophages in vitro,76 and inhibits IL-6 and TNF-α in macrophages77 as well as in whole (human) blood.78 Hypercapnic acidosis reduces TNF-α concentrations in the bronchoalveolar fluid following in vivo pulmonary ischemia-reperfusion.79 Such effects on cytokines and chemokines may be sustained; for example, intraperitoneal macrophages demonstrate impaired TNF-α production for up to 3 days following exposure to hypercapnia.80,81 These actions appear to be mediated, at least in part by inhibition of the transcriptional regulator nuclear factor kappa B (NF-κB).82
Figure 14-4
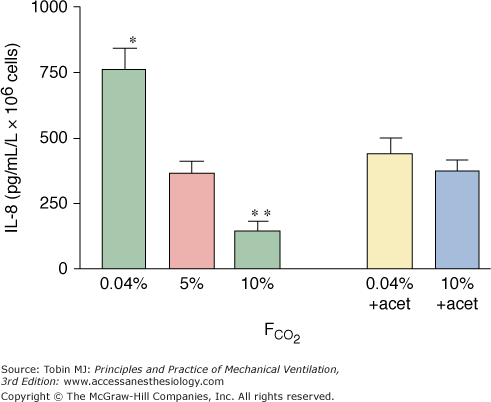
Hypercapnia (10% CO2) inhibits, whereas hypocapnia (0.04% CO2) potentiates, the release of interleukin (IL)-8 from endotoxin-stimulated neutrophils. Incubation with acetazolamide, which impairs the effect of extracellular CO2 on intracellular pH, abolished the influence of extracellular CO2. (Reproduced, with permission, from Coakley RJ, Taggart C, Greene C, McElvaney NG, O’Neill SJ. Ambient pCO2 modulates intracellular pH, intracellular oxidant generation, and interleukin-8 secretion in human neutrophils. J Leukoc Biol. 2002;71:603–610.)
Hypercapnia may impact the cellular immune response via several mechanisms, including impaired coordination of cytokine signaling as well as inhibition of neutrophil expression of key inflammatory and adhesion molecules (e.g., chemokines, selectins, and intercellular adhesion molecules).75 Neutrophil chemotaxis may be directly impaired.83 These effects also occur in vivo as lung neutrophil recruitment is inhibited during ventilator-induced84 and endotoxin-induced85 lung injury. Hypercapnic acidosis directly impairs neutrophil86 and macrophage70 phagocytosis, effects that appear to be a function of the acidosis rather than the CO2 tension.87
The cellular and molecular mechanisms underlying the inhibitory effects of hypercapnic acidosis in the neutrophil and macrophage are increasingly understood. Both hypercapnia and acidosis impair neutrophil intracellular pH regulation. Intracellular pH decreases when neutrophils are activated by immune stimuli75 and, where pH is normal, there tends to be a recovery in neutrophil intracellular pH (toward normal). Hypercapnia decreases extracellular and intracellular pH in the local milieu, resulting in a rapid fall in neutrophil cytosolic pH,88 potentially overwhelming the capacity of phagocytes—especially when activated89—to regulate cytosolic pH.
In common with many biologic systems, the enzymes that produce oxidizing free radicals function optimally at physiologic pH. Oxidant generation by both basal and stimulated neutrophils appears to be regulated by ambient CO2 levels, with oxidant generation reduced by hypercapnia and increased by hypocapnia.75 Production of superoxide by stimulated neutrophils in vitro is decreased at acidic pH.90 Hypercapnic acidosis inhibits the generation of oxidants such as superoxide by neutrophils75 and by macrophages.91 Although free radicals such as superoxide have been implicated in the pathogenesis of injury in acute lung injury and/or ARDS, effective phagocyte generation of free radicals is necessary for killing of ingested bacteria. The finding that hypercapnia reduces endotoxin-induced pulmonary oxidant production92 reinforces concerns that neutrophil-mediated bacterial killing might be critically inhibited.
In the brain, hypercapnic acidosis attenuates glutathione depletion and lipid peroxidation,93 which reflect free-radical activity and tissue damage, respectively. In the lung, hypercapnic acidosis reduces free-radical tissue injury following ischemia–reperfusion79 and attenuates the production of the higher oxides of nitric oxide, such as nitrate and nitrite, following both ventilator-induced94 and endotoxin-induced85 injury. Hypercapnic acidosis inhibits injury mediated by xanthine oxidase and directly inhibits the enzyme.95 Interactions between CO2 and free radicals may also have fundamental effects as recent work indicates that hypercapnia may cause signaling, in part, by cellular oxidation.96
Specific concerns exist regarding the potential for hypercapnia to potentiate tissue nitration by peroxynitrite, a potent free radical. Peroxynitrite is produced in vivo largely by the reaction of nitric oxide with superoxide radical and causes tissue damage by oxidizing a variety of biomolecules and by nitrating phenolic amino acid residues in proteins.97 The potential for buffered hypercapnia to promote the formation of nitration products from peroxynitrite has been clearly demonstrated in vitro.98,99 The potential, however, for hypercapnic acidosis to promote nitration of lung tissue in vivo depends on the injury. For example, hypercapnic acidosis decreased tissue nitration following pulmonary ischemia–reperfusion,79 but increased nitration following endotoxin exposure85,98,100 (Fig. 14-5).
Figure 14-5
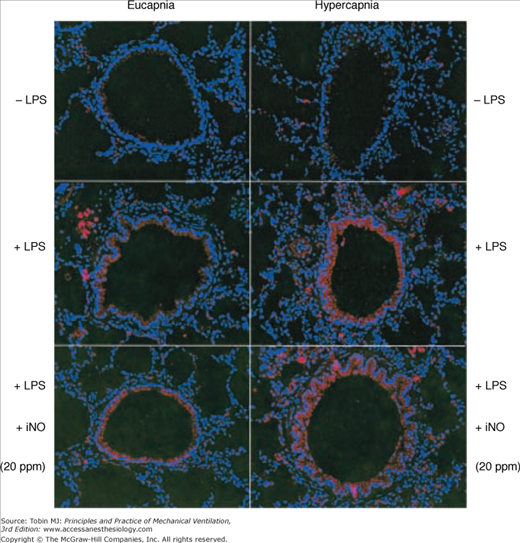
Hypercapnia and inhaled nitric oxide significantly increased the formation of 3-nitrotyrosine following lipopolysaccharide pretreatment. HC, Hypercapnia; iNO, inhaled nitric oxide; LPS, lipopolysaccharide. (Reproduced, with permission, from Lang JD, Figueroa M, Sanders KD, Aslan M, Liu Y, Chumley P, Freeman BA. Hypercapnia via reduced rate and tidal volume contributes to lipopolysaccharide-induced lung injury. Am J Respir Crit Care Med. 2005;171:147–157.)
Some effects of hypercapnia may be mediated by regulation of gene expression. Several unidentified proteins are upregulated by hypercapnia in the normal lung,101 and hypercapnia may regulate this process at the level of gene transcription via altering the half-life of messenger RNA or by modulating protein synthesis. Molecular mechanisms underlying hypercapnic acidosis–mediated control of gene transcription include membrane acid-sensing ion channels and acid-responsive gene promoter regions.102–104 Furthermore, the coding for certain proteins for messenger RNA has pH-sensitive regions.103
Hypercapnic acidosis has been demonstrated to regulate the expression of genes central to the inflammatory response in models of cell injury. NF-κB is a key regulator of the expression of multiple genes involved in inflammatory response, and its activation represents a pivotal early step in activation of the inflammatory response.105 NF-κB is found in the cytoplasm in an inactive form bound to inhibitory proteins called inhibitory protein κB (IκB). The important isoforms are IκB-α and IκB-β. IκB proteins are phosphorylated by the IκB kinase complex and subsequently degraded, thus allowing NF-κB to translocate into the nucleus, bind to specific promoter sites, and activate target genes.105 Hypercapnic acidosis inhibits endotoxin-induced NF-κB activation and DNA binding in pulmonary endothelial cells by decreasing IκB-α degradation82 (Fig. 14-6). Hypercapnic acidosis also suppressed endothelial production of intercellular adhesion molecule-1 and IL-8, which are critically regulated by the NF-κB pathway.82 Importantly, although inhibition of NF-κB may have important antiinflammatory effects, it may also reduce repair following injury.106
Figure 14-6

Hypercapnia suppresses the degradation of IκB-α (A) but not IκB-β (B) following exposure to lipopolysaccharide, thereby inhibiting the nuclear translocation of NF-κB and downstream cytokine production. The effects of isocapnic acidosis and buffered hypercapnia (C) on IκB-α degradation were intermediate between normocapnic control and hypercapnic acidosis conditions. BH, buffered hypercapnia; HA, hypercapnic acidosis; IA, isocapnic acidosis; LPS, lipopolysaccharide; NC, normocapnia; NF-κB, nuclear factor κB. (Reproduced, with permission, from Takeshita K, Suzuki Y, Nishio K, et al. Hypercapnic acidosis attenuates endotoxin-induced nuclear factor-[kappa]B activation. Am J Respir Cell Mol Biol. 2003;29:124–132.)
Hypercapnia appears to inhibit epithelial fluid transport, a potentially important mechanism by which alveolar edema is cleared during lung injury.107 The effect appears to be mediated via hypercapnia per se and seems to be independent of ambient pH.107 The mechanism of action involves adenosine monophosphate (AMP)-activated protein kinase, which, when activated by hypercapnia, promotes Na, K-ATPase endocytosis.108 The decrement in alveolar fluid clearance can be prevented by β-adrenergic agonists or cyclic adenosine monophosphate.108 More recent studies have also implicated extracellular signal-regulated kinase in the activation of AMP-activated protein kinase by hypercapnia, leading to Na, K-ATPase endocytosis.109
Hyercapnia appears to inhibit pulmonary epithelial-cell resealing110 following ventilator-induced lung injury via a pH-dependent mechanism,111 and also inhibits the closure of pulmonary epithelial wounds via a mechanism that involves reduced activation of NF-κB, which, in turn, inhibits cellular migration.106 These effects of hypercapnia on wound healing appear to be a function of CO2 tension rather than acidosis.
Effects in the Setting of Organ Injury
Hypercapnic acidosis attenuates the increased lung permeability consequent to free-radical-mediated lung injury (Fig. 14-7).95 Although hypercapnic acidosis reduces xanthine oxidase activity,95 this does not account for all its protective effects.112 Subsequent in vivo studies confirmed and further characterized the protective effects of hypercapnic acidosis in ischemia–reperfusion lung injury. Hypercapnic acidosis preserved lung mechanics, attenuated protein leakage and reduced pulmonary edema in addition to preserving oxygenation in comparison with control conditions following in vivo pulmonary ischemia–reperfusion,79 as well as in lung injury secondary to splanchnic reperfusion.43 Such protective effects of hypercapnic acidosis are not mediated via decreases in pulmonary artery resistance; on the contrary, protection occurred despite elevated pulmonary artery pressures.43
Figure 14-7

Effects of CO2 on ischemia–reperfusion in the isolated perfused lung. Pulmonary microvascular permeability is significantly less following reperfusion in the presence of hypercapnia (25% CO2) compared with normocapnia (5% CO2). Kf,c, pulmonary microvascular filtration coefficient. (Reproduced, with permission, from Shibata K, Cregg N, Engelberts D, et al. Hypercapnic acidosis may attenuate acute lung injury by inhibition of endogenous xanthine oxidase. Am J Respir Crit Care Med. 1998;158(5 Pt 1):1578–1584.)
The direct effects of hypercapnic acidosis in ventilator-induced lung injury have been examined in both ex vivo and in vivo models. The addition of inspired CO2 decreased ventilator-induced lung injury in the isolated rabbit lung,94 the in vivo rabbit84 (Fig. 14-8), and in the in vivo mouse.113 Not all the data are positive. Supplemental CO2 exhibits more modest protective effects in the setting of more clinically relevant tidal stretch. Strand et al114 demonstrated that significant hypercapnic acidosis (mean PaCO2 of 95 mm Hg) was well tolerated in preterm lambs and also appeared to reduce lung injury. In the context of a clinically relevant high VT adult model (VT 12 mL/kg, positive end-expiratory pressure 0 cm H2O), Laffey et al43 reported that hypocapnia was deleterious and conversely that hypercapnic acidosis was protective. In contrast, inspired CO2 did not attenuate lung injury in an atelectasis-prone model that mimics neonatal respiratory distress syndrome.115 Taken together, these findings suggest that while hypercapnic acidosis substantially attenuates injury secondary to excessive stretch, its effects in the context of more clinically relevant lung stretch or, with extensive atelectasis, may be more modest.
Figure 14-8
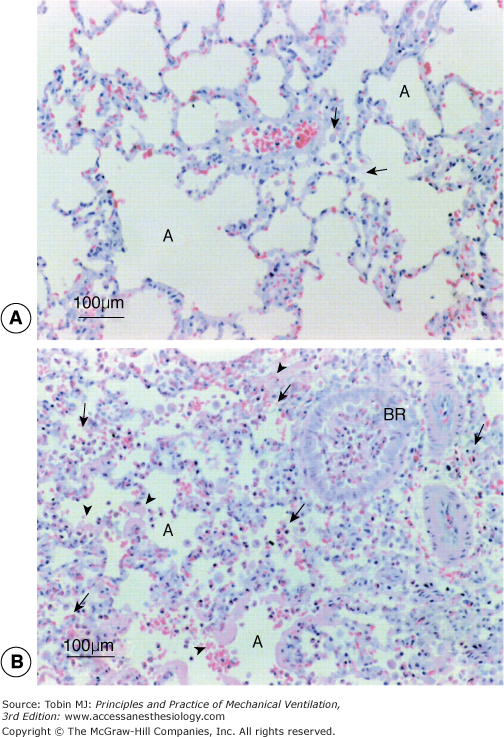
Lung histology following high tidal volume ventilation in the in vivo rabbit. The extent of histologic injury is far less with addition of inspired CO2 (PaCO2 of 80 to 100 mm Hg) (A) than with a PaCO2 of 40 mm Hg (B). (Sinclair SE, Kregenow DA, Lamm WJ, Starr IR, Chi EY, & Hlastala MP. Hypercapnic acidosis is protective in an in vivo model of ventilator-induced lung injury. Am J Respir Crit Care Med. 2002;166:403–408.)
In neonatal models, chronic hypercapnia attenuates the development of hypoxia-induced pulmonary hypertension116,117 in part via augmentation of L-arginase expression resulting in increased local nitric oxide117 and in part via inhibition of endothelin expression.118 Although no mechanism was established, comparable effects were described wherein chronic hypercapnia attenuated the development of long-term hypoxia-induced pulmonary hypertension in the rat.119
The mechanisms of lung injury in sepsis-induced ARDS are quite distinct from those in “sterile” experimental models. Lipopolysaccharide, a key endotoxin of gram-negative bacteria, initiates lung injury by activating a specific receptor, the Toll-like receptor-4.120 Hypercapnic acidosis can reduce the severity of lung injury induced by local administration of endotoxin.85 Mechanisms of sepsis-induced injury, however, include damage to host tissue from the inflammatory response, as well as damage caused by direct bacterial invasion.24,85 In preclinical sepsis models, the effects of hypercapnia appear to depend also on the severity—and phase—of the injury. Although hypercapnic acidosis did not alter the severity of mild injury induced by instilled Escherichia coli,121 it did reduce the severity of more severe lung injury induced by the same organism.122 In the setting of established E. coli pneumonia, short-term hypercapnia reduced lung injury, particularly when effective antimicrobial therapy was used.122 In contrast, in the setting of prolonged E. coli-induced pneumonia (i.e., 72 hours), ongoing environmental hypercapnia worsened the severity of the lung injury.86 Here, prolonged hypercapnia reduced bacterial killing, in part by reduced neutrophil phagocytic activity (Fig. 14-9).86 Subsequent studies confirmed that these effects in prolonged infection appear to be a function of the hypercapnia and not acidosis, as buffered hypercapnia also worsened infection-induced injury.87 Nonetheless, deleterious effects of hypercapnia in the setting of prolonged pneumonia were completely ablated by antimicrobial therapy.86
Figure 14-9

Sustained hypercapnic acidosis worsens pneumonia-induced lung injury and increases bacterial load. Panels A and B represent photomicrographs of section of lung tissue from a lung exposed to normocapnia and hypercapnia respectively, 2 days after intratracheal infection with E. coli. Animals exposed to environmental hypercapnia (inspired CO2 5%) sustained a more severe lung injury. Panel C demonstrates greater bacterial load in lungs from E. coli infected groups exposed to hypercapnia compared to normocapnia. Panel D demonstrates that neutrophils from rats exposed to hypercapnia have a reduced ability to phagocytose fluorescent latex beads compared to neutrophils from normocapnic rats. (Modified, with permission, from O’Croinin DF, Nichol AD, Hopkins N, et al. Sustained hypercapnic acidosis during pulmonary infection increases bacterial load and worsens lung injury. Crit Care Med. 2008;36:2128–2135.)
Concerns regarding harm caused by hypercapnia in prolonged lung infection are reflected in an elegant series of studies in the Drosophilia70 demonstrating an increased mortality in infections with multiple different bacterial pathogens, including Staphylococcus aureus, Enterococcus faecalis, and E. coli.70 The effects appear to be a function of the elevated CO2 (not lowered pH) and are mediated in part via suppression of Rel/NF-κB, an important conserved pathway (Fig. 14-10).70
Figure 14-10
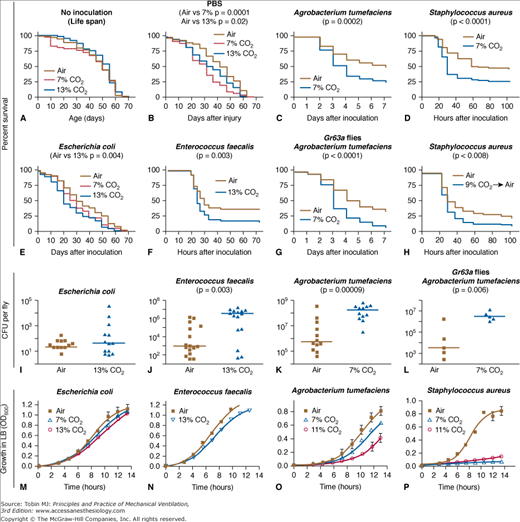
Prolonged hypercapnia decreases resistance of Drosophila to specific bacterial infections. Panel A. Hypercapnia does not affect Drosophila life span. Panels B to H. Hypercapnia slightly increases death of flies inoculated with sterile phosphate-buffered saline (PBS) (Panel B) or with E. coli (Panel E), but significantly increases mortality at CO2 levels as low as 7% after inoculation with A. tumefaciens (Panel C), the human pathogen S. aureus (Panel D), and the Drosophila natural pathogen E. faecalis (Panel F). Immune suppression does not require the neuronal CO2 receptor Gr63a (Panel G). Panel H. Pretreatment of flies with 9% CO2 before S. aureus infection in air is sufficient to increase mortality, even when flies are cultured in air after inoculation. For Panels A to H, unless otherwise noted, flies were exposed to indicated CO2 level for 24 hours before inoculation and returned to hypercapnia until end of assay. We show representative results for the lowest CO2 levels at which significant effects on mortality were consistently observed. Panels I to L. Hypercapnia increases the bacterial load for strains causing increased mortality during hypercapnia. Horizontal lines show medians. Panels M to P. Effects of hypercapnia on bacterial growth. Note that S. aureus growth is dramatically reduced in 7% CO2 even though hypercapnia increases mortality of flies infected with S. aureus. (Reproduced, with permission, from Helenius IT, Krupinski T, Turnbull DW, et al. Elevated CO2 suppresses specific Drosophila innate immune responses and resistance to bacterial infection. Proc Natl Acad Sci U S A. 2009;106(44):18710–18715.)
In a sheep model of established sepsis, hypercapnia augmented hemodynamics (comparably to dopamine) and resulted in increased systemic oxygenation.123 In a small animal model of established systemic sepsis induced by cecal ligation and perforation, hypercapnia had similar effects on hemodynamics.124 In contrast to established pulmonary sepsis,86 hypercapnia reduced the lung injury—in terms of histology and lung mechanics—associated with cecal ligation and perforation124; importantly, in this setting, hypercapnia did not increase bacterial load in the lung, bloodstream, or peritoneal cavity.124 Buffering the hypercapnic acidosis maintained the hemodynamic benefits, but ablated the lung protection.125 Direct intraabdominal administration of carbon dioxide—by introducing operative pneumoperitoneum—may reduce the severity of abdominal sepsis. In experimental endotoxin sepsis, CO2 pneumoperitoneum reduced mortality following laparotomy,126 and insufflation before laparotomy also increased survival.127 These protective effects are seen also in polymicrobial sepsis (Fig. 14-11
Related posts:







Full access? Get Clinical Tree


