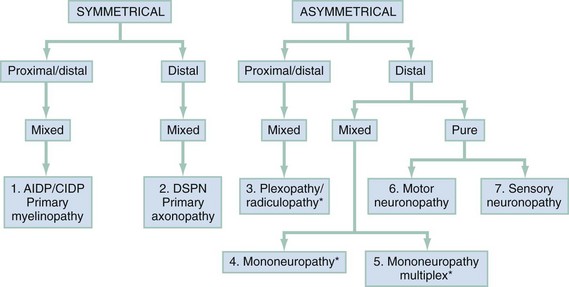
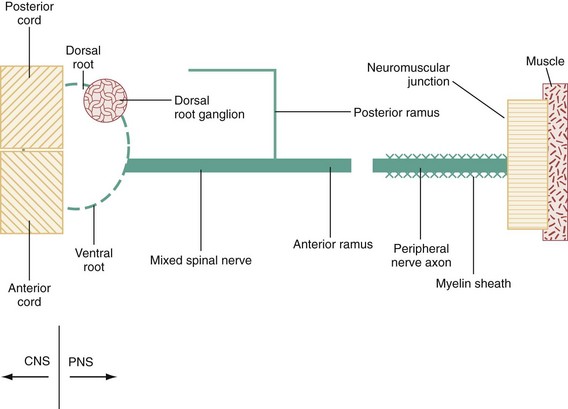
Chapter 107 The nervous system is traditionally divided into central nervous system (CNS) and peripheral nervous system (PNS) components. The PNS can be further subdivided into 12 cranial and 31 spinal nerves. Disorders of the cranial nerves are discussed in Chapter 105. Because diseases of the neuromuscular junction and the myopathies are located distal to the neuron itself, they are also considered separately in Chapter 108. Radiculopathies, which are disorders of the roots of the PNS, are so commonly associated with musculoskeletal neck and back pain that they are mentioned only briefly here and are discussed in detail in Chapter 54. The evaluation of PNS disease involves a goal-directed history and physical examination targeted at answering the following three questions, each of which corresponds to a stratum of the algorithm presented in Figure 107-1: 1. Are the sensorimotor signs and symptoms symmetrical or asymmetrical? 2. Are the sensorimotor signs and symptoms distal or both proximal and distal? 3. Is the modality involved exclusively motor, sensory, or mixed sensorimotor? By systematically combining responses to these questions, one can identify seven discrete categories of peripheral neuropathy, each of which contains a finite set of possible diagnoses. Because pure motor or sensory findings tend to occur mainly in an asymmetrical, distal distribution, this is the only category in Figure 107-1 subdivided into pure motor and pure sensory abnormalities. Although Guillain-Barré syndrome (GBS) is the most commonly encountered emergent peripheral neuropathy in developed countries, its annual incidence is just 1 or 2 cases per 100,000 population.1 In contrast to the acute peripheral neuropathies, several of which are associated with short-term mortality, most peripheral neuropathies seen in the emergency department (ED) are subacute or chronic and are associated not with mortality but with long-term morbidity. Current estimates suggest that about 1.5% of the U.S. population suffers from peripheral neuropathy.2 Diabetes mellitus is a leading contributor. More than 7% of the population has diabetes mellitus, with a prevalence rate of 20% in individuals older than 60 years. Roughly 50% of these individuals develop peripheral neuropathy.3 The spinal component of the PNS is shown schematically in Figure 107-2. The anterior and posterior nerve roots exit the spinal cord at each segmental level. Just distal to the dorsal root ganglion they converge to form a mixed (motor and sensory) spinal nerve, of which there are 31 pairs: 8 cervical, 12 thoracic, 5 lumbar, 5 sacral, and 1 coccygeal. The spinal nerves immediately bifurcate into anterior (ventral) and posterior (dorsal) rami. The posterior ramus travels to the back. The anterior ramus innervates the anterolateral portion of the body and supplies all peripheral nerves for the upper and lower extremities through the brachial and lumbosacral plexus, respectively. Interweaving of fibers occurs within a plexus, producing a mixed sensorimotor innervation of peripheral nerves exiting the plexus. The PNS has only three basic responses to a wide array of pathologic stimuli. As shown in Figure 107-2, these are (1) the myelinopathies, in which the primary site of involvement is limited to the myelin sheath surrounding the axon; (2) the axonopathies, in which the primary site of involvement is the axon, with or without secondary demyelination; and (3) the neuronopathies, in which the cell body of the neuron itself is the primary site of involvement, ultimately affecting the entire peripheral nerve. Although overlap occurs, each of these prototypes has a distinctive clinical presentation, electrophysiologic profile, and microscopic appearance. The differential diagnosis for any patient presenting with sensory, motor, or sensorimotor complaints, particularly if they are localized to the extremities, should include a peripheral neuropathy. Within this group, patients with focal weakness are most concerning because they are at greatest risk for respiratory compromise. Box 107-1 lists the causes of acute, emergent weakness that may affect respiration. Although several of the disorders listed are myopathies (see Chapter 108) rather than peripheral neuropathies, they are lumped together because it is important to identify patients at risk for respiratory failure early in the course of evaluation. As soon as the emergent causes of weakness have been excluded, which is possible in the majority of patients, the individuals with focal weakness should be assessed next to exclude CNS disease (e.g., stroke; see Chapter 101). One can then proceed through the systematic approach to peripheral neuropathy outlined in Figure 107-1. Another way to look at the algorithm displayed in Figure 107-1 is shown in Table 107-1, with the distinguishing features of each of the seven peripheral neuropathic patterns described by distribution and modality and represented by a disease prototype. Table 107-1 Patterns and Prototypes of Peripheral Neuropathies GBS is a heterogeneous and unpredictable disorder, with marked variation in latency between antecedent infection and symptom onset. The clinical signs, cadence of disease progression, degree of respiratory compromise, laboratory findings, and time required for convalescence are also highly variable. The most common form of GBS is an acute inflammatory demyelinating polyneuropathy, representing 90% of the cases seen in the United States.1 Less common variants are acute motor axonal neuropathy, acute motor and sensory axonal neuropathy, and the Miller Fisher syndrome. Acute motor axonal neuropathy, which accounts for most of the remaining cases seen in the United States, afflicts those of Asian descent. Miller Fisher syndrome is a rare form of GBS characterized by the triad of ophthalmoplegia, ataxia, and areflexia (Box 107-2).1,4 The majority of patients seek treatment days to weeks after resolution of an upper respiratory or gastrointestinal illness, presenting with progressive, symmetrical distal (and usually to a lesser extent proximal) weakness. Signs and symptoms are usually worse in the lower extremities and are associated with diminution or loss of deep tendon reflexes (DTRs), variable sensory findings, and sparing of the anal sphincter. Up to 32% will have all four extremities affected at the time of presentation, but only 10% will have weakness that begins in the upper extremities.1 The ocular muscles are usually spared. Urinary retention secondary to autonomic dysfunction may occur, contributing to a clinical picture easily mistaken for a spinal cord lesion or conus medullaris syndrome. The most commonly infectious organisms associated with GBS are Campylobacter jejuni (in patients with a history of diarrhea), cytomegalovirus, Epstein-Barr virus, and Mycoplasma pneumoniae. Acute inflammatory demyelinating polyneuropathy is caused in part by macrophage invasion of the myelin sheath. The macrophage is believed to detect antigens in the myelin that are nearly identical to the antigens present on certain infectious organisms.1,5,6 In practice, patients with symmetrical weakness of relatively acute onset, decreased or absent DTRs, and variable degrees of sensory loss are managed as if they have GBS or one of its variants. These patients have a greater risk for respiratory compromise. Conversely, patients with predominantly sensory signs and symptoms are less likely to develop acute respiratory distress and have a more favorable prognosis.7 About half of patients with GBS have autonomic dysfunction, experience a peak of disease severity within a week of onset, have some form of cranial nerve involvement (usually VII), and suffer long-term sequelae of their illness. Nearly one third require ventilatory support. Both the mortality and the recurrence rate are about 3%.8 In addition to electrophysiologic testing, there are three ancillary tests that may be helpful in the diagnosis of GBS. Cerebrospinal fluid (CSF) analysis is useful when it demonstrates the characteristic picture of markedly elevated protein with only a mild pleocytosis. In the clinical setting of suspected GBS, this finding is highly specific. Early in the disease, however, patients may have normal CSF values. Consequently, a normal CSF value cannot be used to exclude GBS because of the limited sensitivity of this test. Specific protein levels in the CSF (neurofilament NfH) have been shown to correlate with outcomes.9 The GBS disability score, which combines age, presence or absence of diarrhea, and a score of the patient’s ability to ambulate independently at 2 weeks, has been shown to be predictive of prognosis at 6 months, particularly related to independent activity.10 Tongue weakness has been found to be associated with the development of respiratory compromise and the need for mechanical ventilation in patients with GBS.11 Management.: Individuals with suspected GBS should have their respiratory function tested. A decrease in forced vital capacity (FVC) has been shown to correlate with the need for intubation in patients with GBS. An FVC of less than 20 mL/kg is associated with pending respiratory failure and the need for intubation, whereas patients with an FVC of more than 40 mL/kg do not usually require intubation.12,13 Likewise, patients with a negative inspiratory force of less than 30 cm H2O are more likely to require mechanical ventilation.13 Other tests, such as the forced expiratory volume in 1 second and peak flow rate, can also be used to assess respiratory function. Patients unable to perform these tests and those with less than 100% of predicted values should have an arterial blood gas sample obtained. Evidence of alveolar hypoventilation (elevated carbon dioxide [PCO2]) in a patient with an unsecured airway requires a level of intensive monitoring that is impractical in many EDs. Therefore, patients with weakness, carbon dioxide retention, or other evidence of early ventilatory failure should be considered for early, prophylactic intubation.14 Among patients with possible GBS who have normal pulmonary function, extensor neck strength can be monitored to predict impending ventilatory failure. Patients with probable GBS should receive neurologic consultation and admission for treatment with either plasma exchange or intravenous immune globulin (IVIG).There is sound evidence that both of these treatments are superior to placebo and that combination or sequential therapy confers no therapeutic advantage over either intervention alone. Plasma exchange is cumbersome and not available at many hospitals. IVIG is more readily available and is usually administered in a dose of 400 mg/kg/day for 5 days. However, IVIG is expensive, costing roughly $50 to $80 per gram.15,16 Although IVIG is not formally approved by the Food and Drug Administration for this purpose, its use is supported in national treatment guidelines.17 Corticosteroids are no longer recommended for treatment of GBS.18 Oral steroids have been shown to delay recovery. Intravenous steroids alone have been shown to impart no benefit, and although the combination of intravenous steroids and IVIG has hastened recovery, there was no effect on long-term outcome.18–21 The marked elevation in blood pressure seen in some patients with GBS should not be treated because it is typically transient and may be followed by precipitous and unpredictable hypotension. Compared with adults, children who have GBS have neuropathic pain more often (80%) and require mechanical ventilation less often (13%).16, 22 Most polyneuropathies are characterized by a pattern of distal, symmetrical sensorimotor findings, worse in the lower than in the upper extremities, with a stocking-glove distribution of sensory abnormalities that gradually diminishes as one moves proximally. Motor weakness and loss of DTRs, which lag behind the sensory features, follow a similar pattern of progression from distal to proximal. The diffuse, distal, symmetrical nature of this pattern is most consistent with a toxic-metabolic disease process, as yet unidentified, that causes a length-dependent axonopathy. Distal symmetrical polyneuropathy (DSPN) is the most common type of peripheral neuropathy. Only the most common causes of DSPN are discussed, with a more complete listing of causes shown in Box 107-3. Sensory loss continues to move proximally, and before it reaches the knees, the fingertips are usually involved. DTRs are progressively lost, as is proprioception. If loss of proprioception becomes severe, patients may develop sensory ataxia. As the neuropathy continues to progress, sensory abnormalities ultimately involve all modalities and extend to a diamond-shaped periumbilical area. Far-advanced disease may affect sensation over the skull vertex and facial midline structures. Atrophy and areflexia occur as weakness worsens. Severely impaired patients may be unable to ambulate or to grasp objects. These symptoms have a significant impact on the patient’s quality of life, affecting not only physical functioning but also sleep and emotional and social functioning. Many of these patients display signs of depression or anxiety.23 Polyneuropathies can be difficult to diagnose and are best approached by the performance of electrodiagnostic studies for patients with a constellation of symptoms and signs suggesting a particular neuropathy.24 Management.: As with virtually all peripheral neuropathies, referral is indicated for management of diabetic DSPNs. If discomfort is severe, the etiology of the neuropathy seems likely to be diabetic, and if referral is delayed, it may be necessary to provide the patient with some symptomatic relief. Because treatment of neuropathic pain has traditionally been linked to etiology rather than to an underlying mechanism, the choice of pharmacologic agents is empirical, with substantial practice variation in the United States and worldwide. The costs for patients and health plans are considerable; patients will typically spend more than $1000 per year for pain relief from diabetic DSPN.9 Nonsteroidal anti-inflammatory drugs should not be considered first-line treatment because they have little proven efficacy and a high potential for renal impairment.3 On the basis of placebo-controlled randomized clinical trials, tricyclic antidepressants and anticonvulsants appear to have the best NNTs (number of patients needed to treat to provide at least 50% relief of symptoms in one patient). These are generally in the range of 3 to 5, with confidence intervals whose upper limits reach 10 in some instances.25 Imipramine or amitriptyline may be started at a dose of 25 mg at bedtime (10 mg in elders) and titrated slowly up to a dose of 300 mg. Carbamazepine at a dose of 200 to 400 mg every 8 hours and gabapentin at a dose of 900 to 3600 mg/day are also effective treatments.2,26 Tramadol in two studies has shown an NNT below 5.24 Although tramadol is a mixed opioid, development of dependence in long-term use appears to be uncommon. Tramadol combined with acetaminophen has been found to be as effective as gabapentin in the treatment of painful diabetic neuropathy.27 In a recently published guideline, the following medications were recommended for the treatment of neuropathic pain: gabapentin, opioids, tramadol, and tricyclic antidepressants.3 Pregabalin 150 to 600 mg/day, a more recent treatment option, has a mechanism of action similar to that of gabapentin. Duloxetine, a selective serotonin and norepinephrine reuptake inhibitor, has been found effective at a dose of 60 mg/day.3 Recently, tapentadol ER 100 to 250 mg twice daily was found to provide substantial pain relief in patients with diabetic neuropathy.28 Topical capsaicin provides relief in some patients, but the burning associated with its application has limited its use. Improving glycemic control can prevent, diminish, or reverse early diabetic DSPNs, and even in diabetic patients with severe peripheral neuropathy, tight glycemic control can improve functionality and pain control.29
Peripheral Nerve Disorders
Perspective
Epidemiology
Principles of Disease
Pathophysiology
Clinical Features
TYPE
PATTERN DISTRIBUTION
PROTOTYPICAL DISEASE MODALITIES
1
Proximal and distal, symmetrical, sensorimotor polyneuropathy
GBS
Proximal and distal
Symmetrical
Motor > sensory
2
Distal, symmetrical, sensorimotor polyneuropathy
Diabetic DSPN
Distal
Symmetrical
Sensory > motor
3
Proximal and distal, asymmetrical, sensorimotor neuropathy
Brachial plexopathy
Proximal and distal
Asymmetrical
Sensory and motor
4
Distal, asymmetrical, sensorimotor mononeuropathy
CTS (median mononeuropathy)
Distal
Asymmetrical
Sensory and motor
5
Distal, asymmetrical, sensorimotor mononeuropathy multiplex
Vasculitic mononeuropathy multiplex
Distal
Asymmetrical
Sensory and motor
6
Distal, asymmetrical, pure motor neuronopathy
ALS
Distal
Asymmetrical
Motor
7
Distal, asymmetrical, pure sensory neuronopathy
Pyridoxine toxicity
Distal
Asymmetrical
Sensory
Type 1: Demyelinating Polyneuropathies
Guillain-Barré Syndrome
Type 2: Distal Symmetrical Polyneuropathies
Diabetic Distal Symmetrical Polyneuropathy

Full access? Get Clinical Tree


Peripheral Nerve Disorders
Only gold members can continue reading. Log In or Register to continue




