90 Pathophysiology and Classification of Shock States
 Pathophysiology of Shock
Pathophysiology of Shock
Circulatory shock represents a final common pathway of cardiovascular failure. The mortality rate remains high, particularly for patients in cardiogenic and septic shock, for whom the overall mortality rate approximates 50%.1,2 From a physiologic perspective, circulatory shock can be defined as a syndrome in which tissue perfusion is reduced such that blood flow is inadequate to meet cellular metabolic requirements. Clinical manifestations of shock are those of organ hypoperfusion: altered mental status; cool, clammy extremities; decreased blood pressure; decreased pulses; and oliguria.
Hemodynamic Assessment
In patients with circulatory shock, blood pressure should be monitored using intravascular measurements. Vasoconstriction due to compensatory mechanisms to maintain arterial pressure and the use of pharmacologic agents limits the accuracy of noninvasive measurements. This is particularly true in hypodynamic forms of circulatory failure.3
For most vital organs, autoregulatory and neuronal mechanisms maintain blood flow independent of blood pressure at a mean arterial pressure of 60 to 130 mm Hg.4 At either higher or lower levels of pressure, blood flow becomes linearly dependent on blood pressure. Diseases such as hypertension can shift this relationship and increase the critical level of arterial pressure required for organ perfusion. Similarly, impaired autoregulatory mechanisms present in a variety of pathologic states expand the range of pressure-dependent blood flow.
The level of arterial pressure is not a reliable indicator of circulatory performance and tissue perfusion.5,6 In states of hypodynamic circulatory shock, hypotension is a late marker of critical hypoperfusion. As cardiac output falls, blood pressure is initially maintained by increases in peripheral vascular resistance largely mediated by the sympathoadrenal system, and it is only after these mechanisms have been exhausted that hypotension develops. In this setting, tissue hypoperfusion may be present despite normal levels of blood pressure as blood flow is redirected toward more vital organs.7,8 Conversely, hypotension may exist without evidence of organ hypoperfusion. In some vasodilated states, increases in cardiac output maintain vital organ blood flow despite decreased levels of arterial pressure.
Pulmonary artery wedge pressure and central venous pressure are indirect measures of ventricular preload. These measurements correlate poorly with blood volume, end-diastolic volumes, and fluid responsiveness.9,10 Filling pressures are determined by ventricular compliance, venous return, and systolic function. Factors such as ventricular interactions, positive airway pressure, and intrinsic cardiac disease may decrease ventricular compliance and lead to an overestimation of ventricular preload.9 Echocardiographic techniques can provide a more accurate assessment of ventricular loading conditions, while dynamic indicators such as pulse pressure variation or stroke volume variation may provide greater insight as to fluid responsiveness.11,12
 Classification of Shock
Classification of Shock
Hinshaw and Cox proposed a classification of circulatory shock involving four subsets: hypovolemic, cardiogenic, distributive, and obstructive shock.13 This classification can be simplified into two categories with typical hemodynamic profiles (Table 90-1). The first category is hypodynamic shock, which includes the hypovolemic, cardiogenic, and obstructive shock subsets. The second category, hyperdynamic shock, includes distributive shock.
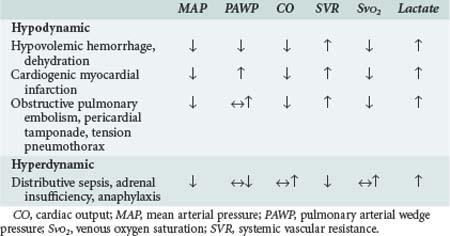
The central features of hypodynamic shock are a low cardiac index and a high-resistance vasoconstricted state. Increased oxygen extraction and lactic acidosis usually parallel the decrease in cardiac output. In cases of hypodynamic shock, the development of organ dysfunction is directly related to inadequate global blood flow. Common causes of hypovolemic shock are hemorrhage, dehydration, and massive capillary leak. Acute decreases in blood volume of 25% result in tachycardia and orthostatic hypotension, whereas decreases of 40% are associated with significant decreases in systolic blood pressure. Decreased filling pressures are the hallmark of hypovolemic shock, in contrast to cardiogenic shock where they are elevated. Acute myocardial infarction involving 40% or more of the ventricular mass is the most common cause of cardiogenic shock.14 Cardiomyopathies and severe valvular lesions are other important causes of cardiogenic shock. Finally, obstructive shock is most commonly due to pericardial tamponade, acute pulmonary embolism, and tension pneumothorax. Since filling pressures are usually increased in these settings (due to outflow obstruction, impaired ventricular filling, and decreased ventricular compliance), distinguishing between obstructive shock and cardiogenic shock can be difficult.
Hyperdynamic circulatory shock is characterized by a high cardiac output and a low-resistance vasodilated state. Filling pressures can be increased or normal depending on volume status and myocardial competence. Common causes of hyperdynamic shock include sepsis, anaphylaxis, some drug intoxications, spinal shock, and adrenal insufficiency. The underlying hemodynamic defect is maldistribution of blood flow and/or blood volume such that effective nutrient blood flow is compromised. In contrast to hypodynamic shock, oxygen extraction may be normal or decreased despite evidence of hypoperfusion.15 Direct mediator-related effects coupled with tissue hypoperfusion produce cellular injury and organ dysfunction in patients with septic shock.
Considerable overlap may exist between these different syndromes. Early in septic and anaphylactic shock, prior to fluid infusion, a significant hypovolemic component usually exists.16 Hypovolemia may be present in a small group of patients presenting with shock due to acute myocardial infarction.17 In the presence of severe sepsis-related myocardial depression, patients with septic shock can develop a hypodynamic profile. Similarly, patients in cardiogenic shock after myocardial infarction and cardiac surgery may demonstrate significant vasodilation due to the activation of mediator cascades while on cardiopulmonary bypass.1,18
Progression of Shock
Critical reductions in tissue perfusion elicit a complex set of reflexes that are directed at maintaining cardiac output and arterial pressure.4 Activation of the sympathetic system increases heart rate and contractility. The release of catecholamines, angiotensin, vasopressin, and endothelins increases arteriolar and venous tone, thereby increasing arterial blood pressure and shifting blood volume from the capacitance vessels to the central circulation. In addition, blood flow is redirected from skeletal muscle, subcutaneous tissue, and the splanchnic circulation to the heart and brain. Vasopressin and activation of the renin-angiotensin system serve to enhance water and sodium retention, thereby protecting intravascular blood volume.
Progression of the shock state is marked by further declines in blood pressure that compromise coronary perfusion and cardiac performance. Increases in peripheral vascular resistance impede left ventricular ejection by increasing left ventricular afterload. Terminal phases of shock are marked by vasomotor dysfunction characterized by loss of arteriolar tone with paradoxical increased venular resistance. The resulting increase in capillary hydrostatic pressure coupled with increased microvascular permeability leads to a loss of intravascular volume and worsening of the shock state. Leukostasis and changes in erythrocyte rheology further impair microvascular blood flow. In animal models of hemorrhagic shock, a state of irreversible shock evolves from which the animals cannot be successfully resuscitated.19
This pathophysiology is altered in patients with hyperdynamic forms of circulatory failure such as septic shock, where inflammatory mediators play a prominent role.20 These patients are characterized by arterial and venous dilation and increased cardiac output. The influence of vasodilatory substances such as nitric oxide predominates over the effects of endogenous and exogenous vasopressor substances. In some forms of vasodilatory shock, inappropriately low levels of vasopressin and cortisol may contribute to vasodilation and refractoriness to catecholamines.21,22 Decreases in capillary cross-sectional area due to the interactions of activated leukocytes, platelets, endothelial cells, and the clotting cascade limit effective nutrient blood flow despite the increase in cardiac output.23,24 Progressive hypotension refractory to fluid infusion and vasopressors results in worsening tissue hypoperfusion, acidosis, and organ failure. A hypodynamic circulation develops as a terminal event.
Oxidative Metabolism in Shock
The primary metabolic defect in circulatory shock is impaired oxidative metabolism with resulting cellular and organ failure. This impairment is most commonly due to decreases in tissue oxygen supply caused by either global decreases in blood flow or maldistribution of blood flow on a regional or microcirculatory level. Systemic oxygen consumption may initially be increased yet inadequate to meet tissue metabolic requirements; however, the terminal phases of all forms of shock are characterized by decreases in oxygen consumption. In experimental studies, the risk of mortality is directly related to the total amount of accumulated oxygen debt.25
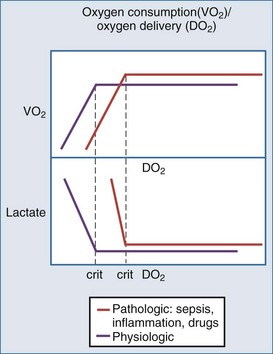
Oxygen delivery is determined by cardiac output, hemoglobin concentration, and the arterial oxygen saturation. Under normal circumstances, oxygen consumption is independent of oxygen delivery and cardiac output (Figure 90-1). Increases in cellular oxygen extraction from a normal level of 25% to a maximum of level of 80% maintain oxygen consumption as blood flow is reduced. When oxygen extraction is maximized, a critical level of oxygen delivery (DO2crit) is reached below which oxygen consumption decreases and anaerobic metabolism ensues. Alterations in vasomotor reflexes due to sepsis or drugs limit maximal oxygen extraction, resulting in critical tissue hypoxia and anaerobic metabolism at higher levels of oxygen delivery.26,27
Aerobic adenosine triphosphate (ATP) generation is dependent on glycolysis occurring in the cytoplasm and oxidative phosphorylation occurring in the mitochondria (Figure 90-2). Under anaerobic conditions, ATP generation is limited to the two ATP generated in the cytoplasm, as compared to the 38 ATP generated aerobically. The decreased entry of pyruvate into the citric acid cycle results in the accumulation of lactic acid and the generation of additional hydrogen ions from the hydrolysis of ATP. Accordingly, the presence of lactic acidosis serves as an indicator of critical cellular deficits in high-energy phosphate metabolism. The normal level of lactate is 0.4 mEq/L to 1.2 mEq/L; levels greater than 2 mEq/L are associated with an increased mortality rate.28
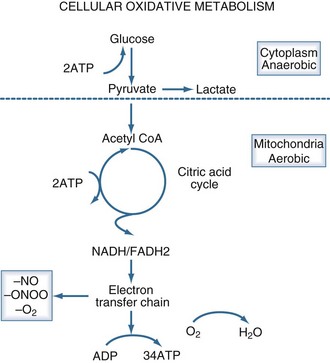
Oxidative metabolism may also be impaired by mechanisms independent of tissue hypoperfusion. A number of inflammatory mediators including nitric oxide, endotoxin, oxygen radicals, calcium, and tumor necrosis factor impair mitochondrial function. Mitochondrial abnormalities have been observed in animal models of septic shock and in cases of reperfusion injury.29 Serum from patients with septic shock inhibits mitochondrial respiration and decreases cellular ATP concentration in vitro.30 A potential pathway of direct mitochondrial impairment involves nitric oxide and its metabolite, peroxynitrite. Both of these substances can directly impair mitochondrial electron chain complexes.31
Accumulation of tissue carbon dioxide (CO2) parallels the development of oxygen debt in circulatory shock.32 Clinically, increases in tissue CO2 levels are manifested by venous hypercapnia and decreases in venous pH. The result is a widening of the arterial-venous CO2 gradient proportional to the degree of circulatory failure. The normal gradient is less than 5 mm Hg, and it can increase to 40 mm Hg during cardiac arrest.33 Decreased clearance of CO2 generated by oxidative processes is responsible for the initial increase in tissue CO2 levels. With the onset of anaerobic metabolism, tissue CO2 excess is largely generated from titration of anaerobically derived acids by bicarbonate. The increase in tissue CO2 levels may have physiologic significance and has been associated with impaired myocardial performance in vitro.



Full access? Get Clinical Tree


