Oxygenation Failure, ARDS, and Acute Lung Injury
KEY POINTS
1 Six mechanisms may contribute to arterial oxygen desaturation: (a) inhalation of a hypoxic gas mixture, (b) alveolar hypoventilation, (c) impaired diffusion of oxygen across the alveolus, (d) 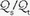 mismatching, (e) shunting of systemic venous blood to the systemic arterial circuit, and (f) abnormal desaturation of systemic venous blood in the presence of a
mismatching, (e) shunting of systemic venous blood to the systemic arterial circuit, and (f) abnormal desaturation of systemic venous blood in the presence of a 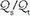 abnormality or shunt.
abnormality or shunt.
mismatching, (e) shunting of systemic venous blood to the systemic arterial circuit, and (f) abnormal desaturation of systemic venous blood in the presence of a abnormality or shunt.2 Clues to the nature of an oxygenation crisis are offered by radiographic appearance. Lung collapse (atelectasis), diffuse or patchy parenchymal infiltration, fluid overload, localized or unilateral infiltration, and a clear chest radiograph are distinct patterns that suggest specific etiologies and approaches to the treatment. Chest CT provides invaluable information to resolve overlapping shadows of lung parenchyma, pleural space, and chest wall.
3 Atelectasis is perhaps the most common cause of hypoxemia for the bedridden, critically ill, and postoperative patient. Potential consequences are worsened gas exchange, pneumonitis, and increased work of breathing. Mobilization, continuous positive airway pressure, and assiduous bronchial hygiene are keys to successful prevention and management.
4 Acute lung injury and acute respiratory distress syndrome are characterized by a delay between a characteristic precipitating event and the onset of dyspnea, impaired respiratory system compliance that results primarily from the loss of functional lung units, markedly reduced aerated lung volume, hypoxemia refractory to modest concentrations of inspired oxygen, and diffuse pulmonary infiltrates. The associated high-protein edema resolves more slowly than hydrostatic edema and is more likely to generate a patchy distribution and detectable air bronchograms.
5 Basic therapeutic principles in treating an oxygenation crisis are to minimize the risk:benefit ratio of ventilation by accepting hypercapnia in preference to high tidal volumes and ventilating pressures, to adequately recruit the lung, and to minimize tissue oxygen demand and ventilation requirements (e.g., by sedation).
6 Shifts of body position alter the regional distributions of ventilation and perfusion and may be associated with changes in tidal and end-expiratory volumes. Prone positioning clearly alters the regional distribution of transpulmonary pressure and may dramatically improve the efficiency of arterial oxygenation associated with an unchanging ventilatory pattern and level of applied positive end-expiratory pressure.
7 Manipulation of peak, mean, and end-expiratory alveolar pressures plays a crucial role in achieving adequate arterial oxygenation at an acceptable FiO2. Moderately high pressures may be needed during a recruiting maneuver to increase the transpulmonary pressure enough to open refractory lung units, especially those in dependent zones. End-expiratory alveolar pressure (total positive end-expiratory pressure, the sum of positive end-expiratory pressure and auto-positive end-expiratory pressure) helps maintain patency of alveolar units at risk for collapse. Under conditions of passive inflation, mean airway pressure reflects average lung size and correlates with oxygenation efficiency.
8 High tidal volume/low positive end-expiratory pressure strategies may extend alveolar injury or retard healing of the already-injured tissues. In the early
phase of acute respiratory distress syndrome, avoiding excessive transpulmonary stretching pressures and inspiratory flow rates during tidal inflation while maintaining sufficient end-expiratory transpulmonary pressure is a rational ventilatory strategy. This approach often results in low tidal volumes (depending on lung compliance) and the need to accept CO2 retention (permissive hypercapnia).
phase of acute respiratory distress syndrome, avoiding excessive transpulmonary stretching pressures and inspiratory flow rates during tidal inflation while maintaining sufficient end-expiratory transpulmonary pressure is a rational ventilatory strategy. This approach often results in low tidal volumes (depending on lung compliance) and the need to accept CO2 retention (permissive hypercapnia).
9 Many choices for ventilatory mode are equally defensible, as long as the practitioner ensures adequate oxygen delivery, follows similar guidelines for lung protection, and remains alert to the potential shortcomings and complications of the mode in use.
10 The essential elements of a lung-protective approach to ventilating acute respiratory distress syndrome are (a) to minimize oxygen and ventilation demands, (b) to apply sufficient end-expiratory and end-inspiratory pressures to maintain nearly complete recruitment of functional alveoli, (c) to avoid overstretching the lung, (d) to accept hypercapnia unless there is a serious neurologic or cardiovascular contraindication, (e) to implement prone positioning in the difficult-to-oxygenate patient, and (f) to consider the potential benefits of avoiding fluid excess and of using adjunctive ventilatory aids (such as inhaled prostacyclin).
▪ OXYGENATION FAILURE
Definitions
Respiratory failure may be considered a problem in one or more of the steps necessary to sustain mitochondrial energy production. Dysfunction may occur in ventilation (the movement of gases between the environment and the lungs; see Chapter 25), in intrapulmonary gas exchange (the process in which mixed-venous blood releases CO2 and becomes oxygenated), in gas transport (the delivery of adequate quantities of oxygenated blood to the metabolizing tissue), or in tissue gas exchange (the extraction or use of O2 and release of CO2 by the peripheral tissues).
The latter two steps in this process may fail independently of the performance of the lung or ventilatory pump. Tissue O2 delivery depends not only on the partial pressure of arterial oxygen (PaO2) but also on the nonpulmonary factors— cardiac output, hemoglobin (Hgb) concentration, and the ability of Hgb to take up and release O2. Cardiogenic shock, severe anemia, and carbon monoxide poisoning provide clinical examples of O2 transport failure (see Chapter 38). Laboratory abnormalities characteristic of such conditions are lactic acidosis and reduced O2 content of mixed-venous blood (even in the face of adequate arterial oxygen tension).
Failure of O2 uptake refers to the inability of the tissue to extract and use O2 for aerobic metabolism. The clearest clinical examples of a derangement in this terminal phase of the oxygen transport chain are septic shock and cyanide poisoning, in which cellular cytochromes (key enzymes in the electron transport process) are inhibited. During sepsis, there is failure of an often generous cardiac output to distribute appropriately and/or an inability of the tissues themselves to make use of the O2 available. Unlike transport insufficiency, failure of tissue uptake implies insufficient O2 extraction and, therefore, may be associated with normal or even high values for mixed-venous oxygen tension, saturation, and content. Some indices that are helpful in other forms of oxygenation failure (i.e., cardiac output, arterial O2 tension, and mixed-venous O2 saturation [SvO2]) may not reflect the impaired tissue O2 uptake reliably; lactic acidosis may be the sole laboratory indicator. Therapy directed at failure of the O2 transport mechanism is discussed in detail elsewhere (see Chapters 1 and 3). The following discussion focuses on the problems that bear on the performance of the lung in oxygenating the arterial blood and in ventilatory failure.
▪ MECHANISMS OF ARTERIAL HYPOXEMIA
Six mechanisms may contribute to arterial oxygen desaturation (Table 24-1):
TABLE 24-1 MECHANISMS OF ARTERIAL HYPOXEMIA | |||||||
|---|---|---|---|---|---|---|---|
|
Inhalation of a hypoxic gas mixture or ascent to altitude
Hypoventilation
Impaired alveolar diffusion of oxygen
Ventilation-perfusion (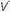 /
/ ) mismatching
) mismatching
Shunting of systemic venous blood to the systemic arterial circuit
Abnormal desaturation of systemic venous blood in conjunction with cause 3, 4, or 5
Low Inspired Oxygen Fraction
A decrease in the partial pressure of inhaled oxygen occurs in toxic fume inhalation, in closed-space fires that consume O2 in combustion, and at high altitudes because of reduced barometric pressure. In this “low fraction of inspired oxygen (FiO2)” context, it should not be forgotten that very rarely, unsuspected disconnection from supplemental oxygen precipitates hypoxemia.
Hypoventilation
Hypoventilation causes the partial pressure of alveolar oxygen (PAO2) to fall when alveolar oxygen is not replenished quickly enough in the face of its ongoing removal by the blood. Although PaO2 may fall much faster than PaCO2 rises during the initial phase of hypoventilation or apnea, the steady-state concentration of PaO2 is predicted by the simplified alveolar gas equation:
In this equation, PiO2 is the partial pressure of the inspired oxygen at the tracheal level (corrected for water vapor pressure at body temperature), and R is the respiratory exchange ratio (i.e., the ratio of CO2 production to oxygen consumption at steady state). Transiently, R can fall to very low values because oxygen is taken up faster than CO2 is delivered to the alveolus. Such a mechanism explains posthyperventilation hypoxemia and may contribute to hypoxemia that accompanies hemodialysis.
Impaired Diffusion
Impaired diffusion of oxygen prevents complete equilibration of alveolar gas with pulmonary capillary blood. Although this mechanism has uncertain clinical importance, many factors that adversely influence diffusion are encountered clinically: increased distance between alveolus and erythrocyte, decreased O2 gradient for diffusion, and shortened transit time of the red cell through the capillary (high cardiac output with limited capillary reserve).
Ventilation-Perfusion Mismatching
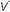 /
/ mismatching is the most frequent contributor to the clinically important O2 desaturation. Lung units that are poorly ventilated in relation to perfusion cause desaturation; high
mismatching is the most frequent contributor to the clinically important O2 desaturation. Lung units that are poorly ventilated in relation to perfusion cause desaturation; high 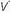 /
/ units contribute to physiologic dead space but not to hypoxemia. The relationship of O2 content to PaO2 is curvilinear. At normal barometric pressure, little additional O2 can be loaded onto blood with already saturated Hgb, no matter how high the O2 tension in the overventilated alveolus may rise. Because samples of blood exiting from different lung units mix gas contents (not partial pressures), overventilating some units in an attempt to compensate for others that are underventilated does not maintain PaO2 at a normal level. Therefore, when equal volumes of blood from well-ventilated and poorly ventilated units mix, the blended sample will have O2 content halfway between them but PaO2 disproportionately weighted toward that of the lower
units contribute to physiologic dead space but not to hypoxemia. The relationship of O2 content to PaO2 is curvilinear. At normal barometric pressure, little additional O2 can be loaded onto blood with already saturated Hgb, no matter how high the O2 tension in the overventilated alveolus may rise. Because samples of blood exiting from different lung units mix gas contents (not partial pressures), overventilating some units in an attempt to compensate for others that are underventilated does not maintain PaO2 at a normal level. Therefore, when equal volumes of blood from well-ventilated and poorly ventilated units mix, the blended sample will have O2 content halfway between them but PaO2 disproportionately weighted toward that of the lower 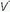 /
/ unit. Even though minute ventilation (VE) and cardiac output (Q) may be absolutely normal, regional
unit. Even though minute ventilation (VE) and cardiac output (Q) may be absolutely normal, regional 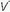 /
/ mismatching will cause PaO2 to fall.
mismatching will cause PaO2 to fall.A high concentration of inspired O2 will correct hypoxemia when 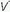 /
/ mismatching, hypoventilation, or diffusion impairment is the cause. (The PAO2 of even poorly ventilated nonshunt units climbs high enough to achieve full saturation.) After breathing 100% O2 for a sufficient period of time, only perfused units that are totally unventilated (shunt units) contribute to hypoxemia. However, when hypoxemia is caused by alveolar units with very low
mismatching, hypoventilation, or diffusion impairment is the cause. (The PAO2 of even poorly ventilated nonshunt units climbs high enough to achieve full saturation.) After breathing 100% O2 for a sufficient period of time, only perfused units that are totally unventilated (shunt units) contribute to hypoxemia. However, when hypoxemia is caused by alveolar units with very low 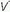 /
/ ratios, relatively concentrated O2 mixtures must be given before a substantial change in the PaO2 is observed (see Fig. 5-2 in Chapter 5).
ratios, relatively concentrated O2 mixtures must be given before a substantial change in the PaO2 is observed (see Fig. 5-2 in Chapter 5).
/ mismatching, hypoventilation, or diffusion impairment is the cause. (The PAO2 of even poorly ventilated nonshunt units climbs high enough to achieve full saturation.) After breathing 100% O2 for a sufficient period of time, only perfused units that are totally unventilated (shunt units) contribute to hypoxemia. However, when hypoxemia is caused by alveolar units with very low / ratios, relatively concentrated O2 mixtures must be given before a substantial change in the PaO2 is observed (see Fig. 5-2 in Chapter 5).Shunting
The term shunt refers to the percentage of the total systemic venous blood flow that bypasses the gasexchanging membrane and transfers venous blood unaltered to the systemic arterial system. Changes in FiO2—either upward or downward—have very little influence on PaO2 when the true shunt fraction, as measured on pure oxygen, exceeds 30% (Fig. 24-1). In contrast, venous admixture of similar magnitude is variably responsive, to the extent
that low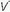 /
/ units account for the hypoxemia. Shunt can be cardiovascular, as in cyanotic right-to-left congenital heart disease, the opening of a patent foramen ovale due to right ventricular overload, or the passage of blood through abnormal vascular channels within the lung (pulmonary arteriovenous malformations). However, by far the most common cause of shunting is pulmonary disease characterized by unventilated alveolar spaces that cannot respond to oxygen therapy. After an extended exposure to an FiO2 of 1.0, all alveoli that remain open are filled with pure oxygen. (Some absorption atelectasis may occur in very low
units account for the hypoxemia. Shunt can be cardiovascular, as in cyanotic right-to-left congenital heart disease, the opening of a patent foramen ovale due to right ventricular overload, or the passage of blood through abnormal vascular channels within the lung (pulmonary arteriovenous malformations). However, by far the most common cause of shunting is pulmonary disease characterized by unventilated alveolar spaces that cannot respond to oxygen therapy. After an extended exposure to an FiO2 of 1.0, all alveoli that remain open are filled with pure oxygen. (Some absorption atelectasis may occur in very low 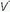 /
/ areas when pure oxygen is breathed, adding to the measured shunt. In the clinical setting, however, the magnitude of this artifact usually is small.) The fraction of blood shunted across the lung from all sources (
areas when pure oxygen is breathed, adding to the measured shunt. In the clinical setting, however, the magnitude of this artifact usually is small.) The fraction of blood shunted across the lung from all sources (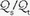 ) can be calculated from the following formula:
) can be calculated from the following formula:
that low
/ units account for the hypoxemia. Shunt can be cardiovascular, as in cyanotic right-to-left congenital heart disease, the opening of a patent foramen ovale due to right ventricular overload, or the passage of blood through abnormal vascular channels within the lung (pulmonary arteriovenous malformations). However, by far the most common cause of shunting is pulmonary disease characterized by unventilated alveolar spaces that cannot respond to oxygen therapy. After an extended exposure to an FiO2 of 1.0, all alveoli that remain open are filled with pure oxygen. (Some absorption atelectasis may occur in very low / areas when pure oxygen is breathed, adding to the measured shunt. In the clinical setting, however, the magnitude of this artifact usually is small.) The fraction of blood shunted across the lung from all sources () can be calculated from the following formula:In this equation, C denotes content, and the lower case letters c, a, and v denote end-capillary, arterial, and mixed-venous blood, respectively. In making such calculations, end-capillary and calculated alveolar oxygen tensions are assumed equivalent. For a patient breathing pure O2, shunt percentages lower than 25% can be estimated rapidly by dividing the alveolar to arterial O2 tension difference (approx. 670—PaO2) by 20, assuming also that the PaCO2 and CvO2 are normal. For example, if measured PaO2 is 270 mm Hg, estimated shunt is 400/20, or 20%.
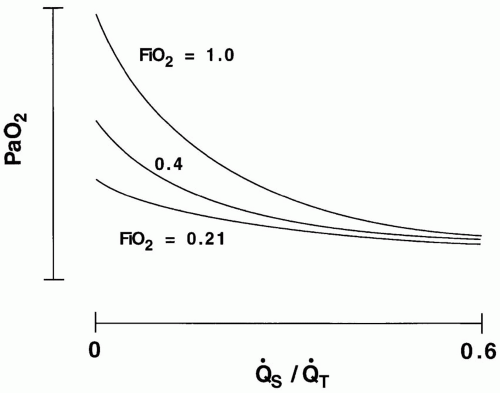 ▪ FIGURE 24-1 Relationship of arterial oxygen tension (PaO2) to true shunt fraction ([Q with dot above] S/[Q with dot above]T) for three values of inspired oxygen fraction (FiO2). Variations of FiO2 exert negligible effects on PaO2 when true shunt exceeds 30%. |
At inspired oxygen fractions lower than 1.0, true shunt cannot be estimated reliably by an analysis of oxygen contents, but “venous admixture” or “physiologic shunt” can. (Many publications erroneously refer to venous admixture from all causes as “shunt.”) Any degree of arterial O2 desaturation can be considered as if it all originated from true shunt units. To calculate venous admixture, CcO2 in the shunt formula is estimated from the ideal alveolar PO2 at that particular fraction of inspired oxygen (FiO2).
Many indices have been devised in an attempt to characterize the efficacy of oxygen exchange across the full spectrum of FiO2 values. Although no index is completely successful, the PaO2:PAO2 ratio and the alveolar to arterial oxygen tension difference (A-a)O2 are often used (see Chapter 5). Both, however, are affected by changes in S[V with bar above]O2, even when the lung tissue itself retains normal ability to transfer oxygen to the blood. Another imprecise but commonly used indicator of oxygen exchange is the PaO2:FiO2 ratio (the P:F ratio). In healthy adults, this ratio normally exceeds 400, whatever the FiO2 may be. Hypoventilation and changes in the inspired O2 concentration minimally alter these ratios in the absence of FiO2-related absorption atelectasis or cardiovascular adjustments.
Abnormal Desaturation of Systemic Venous Blood
The admixture of abnormally desaturated venous blood is an important mechanism acting to lower the PaO2 in patients with impaired pulmonary gas exchange and reduced cardiac output. CvO2, the product of Hgb concentration and S[V with bar above]O2, is influenced by cardiac output (Q), arterial oxygen saturation (SaO2), and oxygen consumption (VO2):
It is clear from this equation that S[V with bar above]O2 is directly influenced by any imbalance between VO2 and oxygen delivery. Thus, anemia that is inadequately compensated by an increase in cardiac output or a cardiac output too low for metabolic needs in a nonanemic patient can cause both S[V with bar above]O2 and PaO2 to fall when the venous admixture percentage is abnormal.
Fluctuations in S[V with bar above]O2 exert a more profound influence on PaO2 when the shunt is fixed, as in regional lung diseases (e.g., atelectasis), than when the shunt varies with changing cardiac output, as it tends to do in diffuse lung injury (acute respiratory distress syndrome [ARDS]) (Fig. 24-2). Even when S[V with bar above]O2 is abnormally low, PaO2 will remain
unaffected if all mixed-venous blood gains access to well-oxygenated, well-ventilated alveoli. (A marked decline in S[V with bar above]O2 without arterial hypoxemia routinely occurs during heavy exercise in healthy subjects.) Therefore, abnormal [V with bar above] matching or shunt is necessary for venous desaturation to contribute to hypoxemia.
unaffected if all mixed-venous blood gains access to well-oxygenated, well-ventilated alveoli. (A marked decline in S[V with bar above]O2 without arterial hypoxemia routinely occurs during heavy exercise in healthy subjects.) Therefore, abnormal [V with bar above] matching or shunt is necessary for venous desaturation to contribute to hypoxemia.
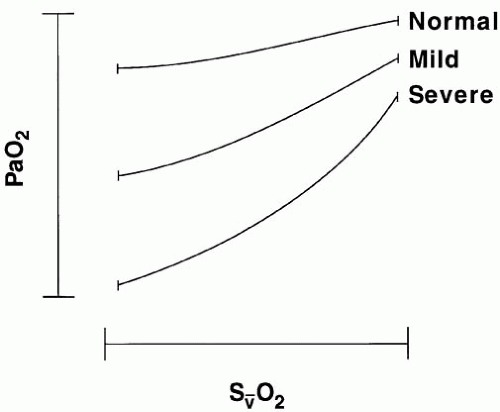 ▪ FIGURE 24-2 Influence of mixed venous oxygen saturation (S[V with bar above]O2) on PaO2 in patients with mild and severe lung disease. Variations in S[V with bar above]O2 related to an oxygen consumption/delivery imbalance have minimal effects on PaO2 in normal subjects but may profoundly affect PaO2 in patients with extensive lung disease. |
▪ DISEASE-INDUCED HYPOXEMIA
Oxygenation disorders can be categorized by their radiographic appearances, which give important clues to the appropriate management approach. Lung collapse (atelectasis), diffuse or patchy parenchymal infiltration, hydrostatic edema, localized or unilateral infiltration, and a clear chest radiograph are common patterns (Fig. 24-3).
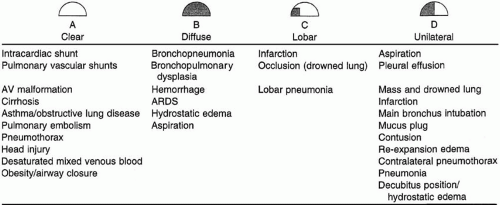 ▪ FIGURE 24-3 Radiographic patterns associated with hypoxemia. |
Atelectasis
Variants of Atelectasis
There are several morphologic types and mechanisms of atelectasis. Regional microatelectasis develops spontaneously in a healthy lung during shallow breathing when it is not periodically stretched beyond its usual tidal range. Platelike atelectasis may be an exaggeration of this phenomenon because of regional hypodistention (e.g., secondary to pleural effusion or impaired diaphragmatic excursion). Both microatelectasis and plate like atelectasis occur most commonly in dependent regions. Lobar collapse usually results from gas absorption behind an airway plugged by retained secretions, a misplaced endotracheal tube, bronchial compression by the heart or pleural effusion, or a large airway mass. Microatelectasis and platelike atelectasis occur routinely in patients on prolonged, uninterrupted bedrest and in postoperative patients who have undergone upper abdominal incisions.
Potential consequences of acute atelectasis are worsened gas exchange, pneumonitis, and increased work of breathing. PaO2 drops precipitously to its nadir within minutes to hours of sudden bronchial occlusion, but it then improves steadily over hours to days as hypoxic vasoconstriction and mechanical factors increase pulmonary vascular resistance through the affected area. Whether an individual patient manifests hypoxemia depends heavily on the vigor of the hypoxic vasoconstrictive response, the abruptness of collapse, and the tissue volume involved. If small areas of atelectasis develop slowly, hypoxemia may never surface as a clinical problem.
Diffuse microatelectasis may be radiographically silent but detectable on physical examination by
dependent (posterior or basilar) end-inspiratory rales, which improve after several sustained deep breaths (sighs) or coughs. Plate atelectasis yields similar physical findings plus tubular breath sounds and egophony over the involved area. Lobar atelectasis gives a dull percussion note and diminished breath sounds if the bronchus is occluded by secretions, but tubular breath sounds and egophony are heard if the central airway is patent. (The latter findings correlate well with the presence of air bronchograms on chest radiograph.) Plate atelectasis develops most frequently at the lung base above a pleural effusion or above a raised, splinted, or immobile hemidiaphragm. Obesity predisposes to all forms of atelectasis. Lobar atelectasis occurs most commonly in patients with copious airway secretions and limited power to expel them. Acute upper lobe collapse occurs less commonly and tends to resolve quickly because of comparatively good gravitational drainage and greater local transpulmonary pressure. Collapse of the left lower lobe is more frequent than collapse of the right lower lobe, perhaps because of its retrocardiac position and its smaller caliber, sharply angulated bronchus. Lobar atelectasis may be complete or partial, but in either case, it is radiographically recognized by opacification in an anatomically geographic distribution, displaced fissures and hilum, compensatory hyperinflation of surrounding tissue, narrowed rib interspaces, and obliterated air/soft tissue boundaries (see Chapter 11). Small amounts of pleural fluid form as an expected consequence of lobar collapse and do not necessarily signify an additional pathologic process.
dependent (posterior or basilar) end-inspiratory rales, which improve after several sustained deep breaths (sighs) or coughs. Plate atelectasis yields similar physical findings plus tubular breath sounds and egophony over the involved area. Lobar atelectasis gives a dull percussion note and diminished breath sounds if the bronchus is occluded by secretions, but tubular breath sounds and egophony are heard if the central airway is patent. (The latter findings correlate well with the presence of air bronchograms on chest radiograph.) Plate atelectasis develops most frequently at the lung base above a pleural effusion or above a raised, splinted, or immobile hemidiaphragm. Obesity predisposes to all forms of atelectasis. Lobar atelectasis occurs most commonly in patients with copious airway secretions and limited power to expel them. Acute upper lobe collapse occurs less commonly and tends to resolve quickly because of comparatively good gravitational drainage and greater local transpulmonary pressure. Collapse of the left lower lobe is more frequent than collapse of the right lower lobe, perhaps because of its retrocardiac position and its smaller caliber, sharply angulated bronchus. Lobar atelectasis may be complete or partial, but in either case, it is radiographically recognized by opacification in an anatomically geographic distribution, displaced fissures and hilum, compensatory hyperinflation of surrounding tissue, narrowed rib interspaces, and obliterated air/soft tissue boundaries (see Chapter 11). Small amounts of pleural fluid form as an expected consequence of lobar collapse and do not necessarily signify an additional pathologic process.
Management of Atelectasis
Prophylaxis
Effective prevention of atelectasis in high-risk patients counteracts shallow breathing, maintains adequate transpulmonary pressure by appropriate positioning or airway pressure, and avoids secretion retention. Obesity, chronic bronchitis, impaired airway clearance, neuromuscular weakness, regional chest wall trauma, recent thoracic or abdominal surgery, and advanced age are predisposing factors. Atelectasis is to be expected whenever the patient is prevented from taking a deep breath by pain, splinting, or weakness. Upper abdominal, lateral chest, midline chest, and lower abdominal incisions are associated with the highest incidence of postoperative atelectasis (in that order). Preoperatively, the airways should be maximally dilated and free of infection. Postoperatively, patients should be encouraged to breathe deeply, to sit upright, and to cough vigorously. Pain should be relieved, but alertness should be preserved. Drainage of excess pleural fluid or ascites deserves consideration. Frequent turning and early mobilization are among the most important prophylactic measures. Continuous positive airway pressure (CPAP) may be helpful, especially for intubated patients. Respiratory therapy (RT) techniques such as airway suctioning, incentive spirometry, and chest vibropercussion (if tolerated) are prophylactically as well as therapeutically effective in well-selected patients (see Chapter 18).
Treatment
Whenever possible, mobilization is a highly effective treatment. Periodic deep breathing effectively reverses platelike atelectasis and microatelectasis. Sustained deep breathing is particularly useful. Whether a higher lung volume is achieved by positive airway pressure or by negative pleural pressure is immaterial, assuming that a similar extent and distribution of distention occurs in both cases. Adequate PEEP or CPAP is accepted as routine in treatment of established collapse. Relief of chest wall pain helps reduce splinting and enables more effective coughing. Intercostal nerve blocks with anesthetic agents such as bupivacaine may be effective for 8 to 12 h. Epidural narcotics also may be effective in certain settings. Retained secretions must be dislodged from the central airways. For the unintubated patient, effective bronchial hygiene is inconsistently accomplished with blind tracheal suctioning alone. Nasopharyngeal airways certainly help, but they are not well tolerated by patients who are awake and are not intended for extended care (see Chapter 6). Vigorous RT initiated soon after the onset of lobar collapse (which may include postural maneuvers and/or vibropercussion) can reverse most cases of atelectasis that are due to airway plugging within 24 to 48 h. As a rule, fiberoptic bronchoscopy should be reserved for patients with symptomatic lobar collapse who lack central “air bronchograms” that branch into the collapsed zone and who cannot undergo (or fail to respond to or tolerate) 48 h of vigorous RT. Even whole lung collapse usually merits at least one RT treatment (including stimulated cough and tracheal suctioning) before bronchoscopy is performed. After re-expansion, a prophylactic positioning and RT program should be initiated to prevent recurrence. Adjunctive measures (e.g., bronchodilators, hydration, and
frequent turning and in some cases, mucolytics and mucus lubricants) should not be ignored.
frequent turning and in some cases, mucolytics and mucus lubricants) should not be ignored.
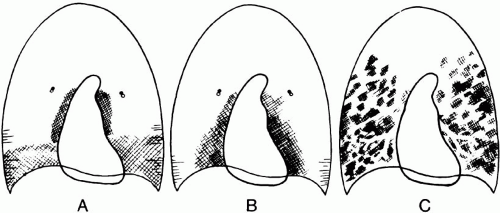 ▪ FIGURE 24-4 Radiographic patterns in patients with impaired oxygenation because of congestive heart failure (A), vascular congestion because of volume overload (B), and ARDS (C). Kerley lines, widened vascular pedicle, costophrenic angle sparing, blurred hilar structures, and paucity of air bronchograms help distinguish congestive heart failure from ARDS. (Reprinted from Milne et al. Am J Roentgenol. 1985;144:879-894, with permission.) |
Diffuse Pulmonary Infiltration
Fluid confined to the interstitial spaces may cause hypoxemia as a result of peribronchial edema,  /
/ mismatching, and microatelectasis; however, very few processes are confined exclusively to the air spaces or to the interstitium. Radiographic signs of extensive alveolar filling include segmental distribution, coalescence, fluffy margins, air bronchograms, rosette patterns, and silhouetting of normal structures. A diffuse infiltrate is said to be largely “interstitial” if these signs are largely absent and the infiltrate parallels the vascular distribution. Computed tomography imaging greatly increases the diagnostic precision, especially when a thickened or diseased chest wall obscures radiographic features of the lung parenchyma. Any diffuse interstitial process will appear more radiodense at the bases than at the apices, in part because there is more tissue to penetrate and because vascular engorgement tends to be greater there. Gravitationally dependent alveoli also are less distended, so the ratio of aerated volume to total tissue volume declines.
mismatching, and microatelectasis; however, very few processes are confined exclusively to the air spaces or to the interstitium. Radiographic signs of extensive alveolar filling include segmental distribution, coalescence, fluffy margins, air bronchograms, rosette patterns, and silhouetting of normal structures. A diffuse infiltrate is said to be largely “interstitial” if these signs are largely absent and the infiltrate parallels the vascular distribution. Computed tomography imaging greatly increases the diagnostic precision, especially when a thickened or diseased chest wall obscures radiographic features of the lung parenchyma. Any diffuse interstitial process will appear more radiodense at the bases than at the apices, in part because there is more tissue to penetrate and because vascular engorgement tends to be greater there. Gravitationally dependent alveoli also are less distended, so the ratio of aerated volume to total tissue volume declines.
/ mismatching, and microatelectasis; however, very few processes are confined exclusively to the air spaces or to the interstitium. Radiographic signs of extensive alveolar filling include segmental distribution, coalescence, fluffy margins, air bronchograms, rosette patterns, and silhouetting of normal structures. A diffuse infiltrate is said to be largely “interstitial” if these signs are largely absent and the infiltrate parallels the vascular distribution. Computed tomography imaging greatly increases the diagnostic precision, especially when a thickened or diseased chest wall obscures radiographic features of the lung parenchyma. Any diffuse interstitial process will appear more radiodense at the bases than at the apices, in part because there is more tissue to penetrate and because vascular engorgement tends to be greater there. Gravitationally dependent alveoli also are less distended, so the ratio of aerated volume to total tissue volume declines.The major categories of acute disease that produce diffuse pulmonary infiltration and hypoxemia are pneumonitis (infection and aspiration), cardiogenic pulmonary edema, intravascular volume overload, and ARDS. From a radiographic viewpoint, these processes may be difficult to distinguish; however, a few characteristic features are helpful.
Hydrostatic Edema
Perihilar infiltrates (sparing the costophrenic angles), a prominent vascular pattern, and a widened vascular pedicle suggest volume overload or incipient cardiogenic edema (Fig. 24-4). A gravitational distribution of edema is highly consistent with well-established left ventricular failure (or long-standing, severe volume overload), especially when accompanied by cardiomegaly and a widened vascular pedicle. Patchy peripheral infiltrates that lack a gravitational predilection and show reluctance to change with position suggest ARDS. Interestingly, septal (Kerley) lines and distinct peribronchial cuffing are very seldom seen in ARDS without coexisting volume overload (see following). On the other hand, prominent air bronchograms are quite unusual with purely hydrostatic etiologies but occur commonly in permeability edema (ARDS) and pneumonia. It should be recalled that permeable vessels leak fluid even at normal vascular pressures, so mixed patterns of ARDS and hydrostatic edema are often seen.
Variants of Hydrostatic Edema
Hydrostatic pulmonary edema (HPE) may occur in multiple settings that have differing implications for prognosis and treatment. The most familiar form of HPE accompanies left ventricular failure. In this setting, signs of systemic hypoperfusion and inadequate cardiac output often accompany oxygenation failure. However, HPE can develop even with a normally well-compensated ventricle during transient cardiac dysfunction (ischemia, hypertensive crisis, arrhythmias, etc.). When the myocardium fails to fully relax during diastole (diastolic dysfunction), volume loading or temporary disturbances of left heart contractility (e.g., ischemia), mitral valve functioning, or heart rate or rhythm may cause rapid, transient alveolar flooding known as “flash pulmonary edema.” In
this setting, an impressive radiographic appearance may both develop and resolve quickly.
this setting, an impressive radiographic appearance may both develop and resolve quickly.
Acute Lung Injury and ARDS
Definitions and Categories
For many years after its original description, ARDS signified the “adult” respiratory distress syndrome. In current parlance, however, acute lung injury (ALI) is a general term that is applied to all degrees of radiographically apparent, diffuse hypoxemic lung injury of diverse etiologies, with ARDS being the most severe form of ALI—regardless of age. “Acute respiratory distress syndrome” is an imprecise term that is often applied to any acute diffuse parenchymal infiltration associated with severe hypoxemia and not attributable to HPE. Published definitions are quite liberal and do not specify the characteristics of the acute infiltrates, nor the mechanical properties of the thorax, nor the conditions under which the oxygenation data were gathered. Several published studies document that experts frequently disagree on whether a radiograph is consistent with “ARDS,” and others have shown that modifications of PEEP and/or position can move a potential candidate into or out of the accepted PaO2/FiO2 criterion. Given these definitional shortcomings, it may not be surprising that few “positive” therapeutic trials in ALI/ARDS have appeared, even though the mortality of patients with similar severity of physiologic impairment has clearly declined over the past decades. The ARDS designation may be most useful when restricted to acute noncardiogenic pulmonary edema with certain characteristic features:
Brief delay between the precipitating event and rapidly developing dyspnea
Impaired respiratory system compliance
Markedly reduced aerated lung volume
Hypoxemia refractory to modest levels of inspired oxygen and PEEP
Delayed resolution
Primary (Pulmonary) Versus Secondary (Extrapulmonary) Acute Lung Injury
The pathogenesis of permeability edema is almost certain to vary with the inciting event. Despite its many diverse causes, sepsis, pneumonia, aspiration, and multiple traumas account for most cases.
Related posts:







Full access? Get Clinical Tree


