Fig. 1
Muscular mechanical hyperalgesia after exercise and changed expression of NGF mRNA in the muscle of rat. (a) Muscular mechanical hyperalgesia developed after lengthening contraction (LC) of the extensor digitorum muscle (black circle), and it was reversed by intramuscular injection of anti-NGF antibody (10 μg, gray square). ***, +++ p < 0.001 compared with day −1. (b) NGF mRNA was increased 12 h–3 days after LC (white columns, exercised side; black columns, contralateral side) (cited from Murase et al. 2010)
Increased NGF mRNA in exercised muscle was first detected 12 h after LC; that is, up to that time point, NGF mRNA did not change (Fig. 1b). NGF protein also increased 12 h after LC up to 1 day after LC (Murase et al. 2010). In addition, anti-NGF antibody injected intramuscularly 2 days after LC, when the muscular mechanical hyperalgesia was the strongest, clearly reversed the mechanical hyperalgesia in 3 h (Fig. 1a). In situ hybridization showed that increased signals of NGF mRNA were seen around the nuclei of muscle/satellite cells 12 h after LC (Murase et al. 2012). Single-fiber recording from muscle C-fibers (sample recording in Fig. 2a, b) showed that NGF 0.8 μM (5 μL) decreased the mechanical threshold of these fibers (Fig. 2c) and increased the discharge number in response to ramp mechanical stimulation (Fig. 2d) (Murase et al. 2010). These changes were observed 10–20 min after injection and lasted as long as the recordings were continued (up to 2 h). This observation differs from that by Mense’s group, which observed no sensitization induced by NGF (Hoheisel et al. 2005), possibly because they recorded only up to 15 min after injection.
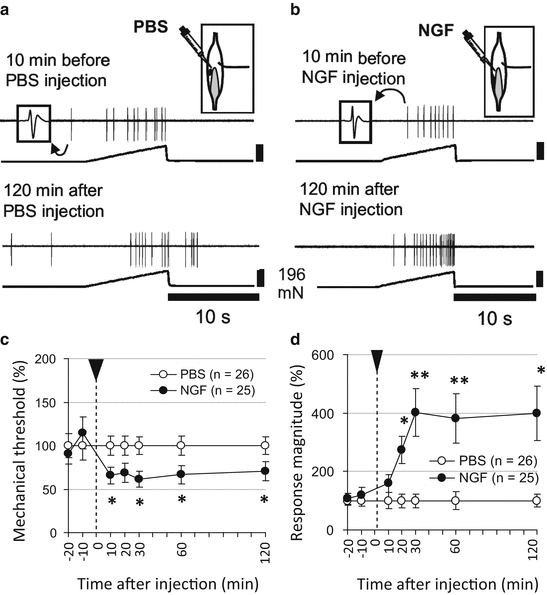
Fig. 2
Sensitization of muscular C-fiber afferents to mechanical stimulation by intramuscularly injected NGF (0.8 μM, 5 μL). (a, b) Sample recordings of C-fiber mechanosensitive afferents from the rat muscle–nerve preparation. (c) Change in the mechanical threshold; (d) number of discharges responding to mechanical stimulation. Values are presented as % of the averaged value of PBS group at each time point. *p < 0.05 and **p < 0.01 compared with PBS group at each time point (cited from Murase et al. 2010)
NGF also facilitates the heat response of muscle C-fibers (Queme et al. 2013). Notably, the heat sensitivity of muscle afferents 2 days after LC was not facilitated in comparison with that recorded from normal muscle (Taguchi et al. 2005b; Queme et al. 2013). This is puzzling because NGF is upregulated in the muscle after LC and NGF can sensitize muscle thin fibers to heat (Queme et al. 2013). This discrepancy might be due to a concentration of NGF in DOMS muscle that is too low to induce heat sensitization or to a substance other than NGF that is involved in the sensitization of nociceptors to mechanical stimulation but not to heat stimulation. We have reported that GDNF is also upregulated in the muscle cells/satellite cells of exercised muscle and implicated in DOMS (Murase et al. 2012, 2013). Our preliminary observation suggests that NGF and GDNF may collaborate in inducing mechanical hyperalgesia.
The upregulation of NGF in the muscle after LC was found to be blocked by HOE 140, a bradykinin B2 receptor antagonist, injected before LC but not after LC. The development of DOMS was also blocked by this procedure (Murase et al. 2010). This observation suggests that a B2 receptor agonist is released during exercise and stimulates NGF upregulation. This B2 agonist is probably Arg-bradykinin in rats, as Boix et al. (2002) have already shown. However, a B2 agonist alone would not seem to be sufficient to induce upregulation of NGF in the muscle, and so some unknown factor(s) is (are) also needed (Murase et al. 2012).
Involvement of TRPV1 in NGF-induced muscular mechanical hyperalgesia has also been shown using capsazepine and TRPV1 knockout mice (Ota et al. 2013). Involvement of ASIC in DOMS was also demonstrated (Fujii et al. 2008).
It is well known that when an LC bout is repeated after days or a few weeks, DOMS after the second bout is much less severe. This is called a repeated bout effect (Chen et al. 2007). NGF upregulation was also found to be reduced after the second LC (Urai et al. 2012).
5.3.2 Role in Cast Immobilization and Osteoarthritis Models
Cast immobilization is known to induce a CRPS 1-like painful condition (Guo et al. 2004; Ohmichi et al. 2012). After experimental cast immobilization of the hind leg, NGF is reported to increase in the hind paw skin (Sekino et al. 2014) and the gastrocnemius muscle (unpublished observation from our lab). NGF upregulation after joint immobilization was also observed in the DRGs (Nishigami et al. 2013). Whether hyperalgesia in these conditions is reduced by anti-NGF antibody or TrkA antagonist has not yet been examined. In addition, the cause of NGF upregulation has also not been explored.
Involvement of NGF in osteoarthritis has been suggested by the observation that NGF is upregulated in osteoarthritic chondrocytes (Iannone et al. 2002) and synovial fluid (Aloe et al. 1992) and pain is alleviated by injection of a soluble NGF receptor, TrkAd5, in a mouse model of osteoarthritis induced by surgical joint destabilization (McNamee et al. 2010). In the latter model, NGF was upregulated in the joints during both postoperative (day 3) and OA (16 weeks) pain phases, but not in the non-painful stage of disease (8 weeks post-surgery). The important role of NGF in OA pain has been indicated by the high effectiveness of TrkAd5 in suppressing pain in both painful phases (McNamee et al. 2010).
Inflammatory processes do not seem to be involved in the abovementioned hyperalgesic conditions; therefore, NGF upregulation might be induced by another mechanism, for example, muscle contraction in the case of DOMS.
5.4 Role of NGF in Visceral Painful Conditions
Upregulation of NGF and its high affinity receptor TrkA was shown in samples obtained from inflammatory bowel disease patients (Crohn’s disease and ulcerative colitis) (di Mola et al. 2000). Cells expressing NGF and TrkA are reported to be polymorphonuclear-like cells of the lamina propria, mast cells, and some ganglionic cells.
There are also conditions where no apparent inflammation is the cause of NGF upregulation. Urinary (Kim et al. 2006) and serum (Jiang et al. 2013) levels of NGF were found to be higher in overactive bladder patients. Because NGF levels were decreased with antimuscarinic and botulinum toxin treatment (Liu et al. 2009), contraction of the bladder smooth muscles seems to be the cause of this upregulation of NGF. Urinary levels of NGF can be a marker for diagnosis of overactive bladder and evaluation of the effects of treatment for it (see Bhide et al. 2013 for review). In addition, when the vector encoding NGF was experimentally infected into the bladder, NGF was upregulated fourfold, and bladder overactivity was induced without any histological evidence of inflammation (Lamb et al. 2004). Collectively, contraction of the bladder and NGF upregulation seem to develop into a vicious cycle. The question of which is the initiating event will be an interesting issue for study.
5.5 Role of NGF in Cancer Pain (and Cachexia) and Other Conditions
All nociceptive, inflammatory, and neuropathic pains can be induced in cancer. Bone metastasis of breast cancer, prostate cancer, and lung cancer induces bone pain. Breakthrough pain induced by movement of involved bone is difficult to treat, and more effective treatments are being sought. A bone metastasis model made by injecting tumor cells into the bone marrow (Halvorson et al. 2005; Bloom et al. 2011) showed that tumors themselves and peripheral bone in the vicinity of the tumor lack detectable innervation, whereas the periosteum is densely innervated in areas where NGF is upregulated. Anti-NGF antibody alleviates pain and decreases the ectopic sprouting of nociceptors into the periosteum (Halvorson et al. 2005; Bloom et al. 2011). NGF blockade decreases tumor proliferation, nociception, and weight loss (Ye et al. 2011).
Administration of a blocking antibody to NGF produced a significant reduction in both early- and late-stage bone cancer pain-related behaviors (Halvorson et al. 2005). Thus, anti-NGF antibody therapy may be particularly effective in blocking bone cancer pain (Pantano et al. 2011). Clinical trials of anti-NGF antibody for the treatment of bone cancer pain still continue.
6 Effect of NGF on Nociceptor Activities and Their Axonal Properties
In normal skin, chronic (10–12 days) deprivation of NGF produced by continuous infusion of TrkA–IgG fusion molecule by an osmotic minipump decreased the percentage of cutaneous nociceptors that responded to heat and/or bradykinin in adult rats but not the percentage that responded to mechanical stimuli (Bennett et al. 1998). This report also showed that the innervation density was reduced in the epidermis with sequestration of NGF. This experiment demonstrates that endogenous NGF in normal adult animals modulates sensitivities to heat and bradykinin but not to mechanical stimuli and the innervation density of terminal axons.
In an inflammatory condition induced by carrageenan, the percentage of spontaneously active fibers recorded in vitro from skin–nerve preparations increased. Sensitivity to heat and bradykinin, but not to mechanical stimulation, also increased. When the TrkA–IgG fusion molecule was coadministered with carrageenan, sensitization to heat and bradykinin of cutaneous nociceptors did not occur, and again, their mechanical sensitivities were not changed (Koltzenburg et al. 1999).
The effects of NGF on muscle nociceptors seem somewhat different from those on skin. Mann et al. (2006) reported that intramuscular injection of human (not rat) NGF into the rat masseter muscle failed to evoke afferent discharges; however, it did decrease the mechanical threshold of masseter A-delta afferent fibers (Mann et al. 2006). Single-fiber recording from C-fibers innervating the extensor digitorum longus (EDL, sample recording in Fig. 2a, b) muscle showed that NGF 0.8 μM (5 μL) decreased their mechanical threshold (Fig. 2c) and increased the discharge number in response to ramp mechanical stimulation (Fig. 2d) (Murase et al. 2010) when compared with fibers recorded from normal rats that received PBS injection.
Axonal properties are also reported to be changed by NGF. Djouhri et al. (2001) reported that NGF sequestration by injecting NGF-binding domain (amino acids 285–413 of TrkAIg2) prevented the following CFA-induced changes in nociceptive neurons with A-delta or C-fibers: increased frequency that a fiber can follow, increased proportions of units with ongoing activity, and decreased action potential duration.
Hirth et al. (2013) also showed by single-fiber recordings 3 weeks after one-time intradermal injection of NGF to pig skin that NGF increased conduction velocity and decreased activity-dependent slowing of mechano-insensitive fibers. They also showed an increase in mechanosensitive fibers and decrease in median mechanical threshold. In contrast to the previous report using continuous infusion of NGF to the rat ankle skin (Bennett et al. 1998), they could not find any increase in the density of intraepidermal nerve fibers (Hirth et al. 2013). The abovementioned changes in axonal properties, especially activity-dependent slowing of conduction velocity, are reported to be related to the availability of Na channels (De Col et al. 2008). The tetrodotoxin-resistant (TTX-r) sodium channels Nav1.8 and Nav1.9 are predominantly expressed in small-/medium-sized nociceptive neurons that are cell bodies of thin-fiber afferents, and increased expression of these channels by NGF has been reported (Fjell et al. 1999; Bielefeldt et al. 2003).
7 Action Mechanism of NGF in Modulating the Nociceptive System
7.1 Mechanism of NGF-Induced Acute Sensitization of Nociceptors to Heat
7.1.1 Direct Phosphorylation by TrkA
TRPV1 is activated by heat (ca. 43 °C) and believed to be involved in heat transduction in nociceptors (Caterina et al. 1997), at least in inflammatory heat hyperalgesia (Caterina et al. 2000; Davis et al. 2000). It was reported that NGF was unable to induce heat hyperalgesia in TRPV1-deficient mice (Chuang et al. 2001). Therefore, the mechanism of the acute sensitizing effect of NGF on the heat response of nociceptors has been often studied using a TRPV1 stimulant, capsaicin, instead of heat.
Shu and Mendell (1999, 2001) first showed that a 10-min application of NGF facilitated capsaicin-induced currents in DRG neurons and later confirmed this observation (Zhu et al. 2004). Even though TrkA is connected with the PLC pathway and an earlier study showed that NGF-induced sensitization was blocked by PKA inhibition (Shu and Mendell 2001), this was not confirmed in later reports (Bonnington and McNaughton 2003; Zhu and Oxford 2007). Activation of protein kinase C by phorbol ester can sensitize the nociceptive neuron response to capsaicin (Bhave et al. 2003), while PKC inhibition abolishes or reduces NGF-induced TRPV1 sensitization (Bonnington and McNaughton 2003; Zhu and Oxford 2007). The effect of inhibiting CaMKII also differed among reports (Bonnington and McNaughton 2003; Zhu and Oxford 2007).
7.1.2 Membrane Trafficking of TRPV1 by TrkA
NGF promotes TRPV1 insertion into the plasma membrane (Zhang et al. 2005), for which involvement of PI3kinase (Stein et al. 2006) and its downstream Src kinase were reported. Src kinase reportedly phosphorylates Tyr200 of TRPV1 and translocates it to the cell membrane (Zhang et al. 2005). The early phase of heat hyperalgesia can be explained by this rapid sensitization (within several min) to heat by NGF.
7.1.3 Indirect Action of NGF Through Degradation of Mast Cells
Previous mast cell degranulation by compound 48/80 or pretreatment with antagonists of 5HT, contained in mast cell granules in rats, reduced the early phase of heat hyperalgesia (or delayed its onset) (Lewin et al. 1994; Amann et al. 1996; Woolf et al. 1996). These observations suggest that NGF also acts indirectly by activating mast cells and neutrophils, which in turn release additional inflammatory mediators causing hypersensitivity to heat.
7.2 Mechanism of NGF-Induced Long-Lasting Sensitization to Heat
The later phase (7 h–4 days after NGF) of heat hyperalgesia appeared to be centrally maintained, since it could be selectively blocked by the noncompetitive NMDA receptor antagonist MK-801 (Lewin et al. 1994). NGF binds to TrkA and is transported to DRG neurons to change the expression of neuropeptides (Donnerer et al. 1992, 1993; Leslie et al. 1995), sodium channels (Fjell et al. 1999), ASIC (Mamet et al. 2002), and other properties. In addition, NGF can increase TRPV1 expression (Donnerer et al. 2005; Xue et al. 2007), via the Ras–mitogen-activated protein kinase pathway (Ji et al. 2002) in DRG neurons. This increased expression of TRPV1 by NGF is implicated in maintaining the heat hyperalgesia in inflammation. Plastic changes in synaptic connections of muscle afferents in the spinal cord have been also reported after long-lasting injection of NGF to the muscle (Lewin et al. 1992).
7.3 Mechanism of NGF-Induced Sensitization to Mechanical Stimuli
The short latency action of NGF on heat sensitivity is well accepted; however, discrepancy exists in the time course of NGF-induced mechanical sensitization. An earlier study showed a latency of 7 h. The shortest latency of sensitization was 10–20 min in single-fiber recording in vitro (Murase et al. 2010) and also in nociceptive behavior although its peak was observed 3 h after injection (Malik-Hall et al. 2005). The longest latency so far reported is 3 days (Hirth et al. 2013). Medium latency of 1 h has been reported (sensation in humans by Svensson et al. 2003, afferent activities by Mann et al. 2006).
Mechanical hypersensitivity several hours after intraplantar injection of NGF was abolished in sympathectomized animals or delayed in mast cell degranulated animals by compound 48/80 (Woolf et al. 1996).
Malik-Hall et al. (2005) reported that acute mechanical hyperalgesia was reduced by inhibitors of the three major pathways for TrkA receptor signaling, extracellular signal-related kinase (ERK)/mitogen-activated protein kinase kinase (MEK), PI3K, and PLCγ. However, inhibitors of kinases downstream of PI3K and PLCγ (glycogen synthetase kinase 3, CAMII-K, or PKC) failed to reduce mechanical hyperalgesia. Thus, they could not clarify the downstream pathways.
Not much cell-based research has been done so far, possibly because the calcium imaging method cannot be applied for the mechanical response or because mechanotransducing channels of nociceptors have not been identified yet. Di Castro et al. (2006) showed that mechanically activated currents in cultured small and IB4(−) neurons was increased after application of NGF for 8 h (not 1 h) through a transcriptional mechanism. The augmented currents were further facilitated by activation of PKC by phorbol ester, and this effect was blocked by tetanus toxin, suggesting that the insertion of new channels into the cell membrane is involved in sensitization (Di Castro et al. 2006). In this report, no early sensitization was observed.
Involvement of TRPV1 was reported in mechanical hyperalgesia after lengthening contraction, where NGF plays a pivotal role (Fujii et al. 2008; Ota et al. 2013). Further research is needed to answer the question of whether mechanisms reported for heat hyperalgesia or augmented response to capsaicin (Bhave et al. 2003; Bonnington and McNaughton 2003; Zhang et al. 2005; Zhu and Oxford 2007) also work in NGF-induced mechanical hyperalgesia.
7.3.1 TrkA or p75NTR
A receptor for NGF that is believed to be involved in heat and mechanical hyperalgesia is TrkA. Recent reports also showed involvement of p75NTR in mechanical hyperalgesia (Iwakura et al. 2010; Khodorova et al. 2013; Matsuura et al. 2013). Downstream signaling cascades are different between these two NGF receptors, and the p75NTR cascade is a sphingomyelin signaling cascade that includes neutral sphingomyelinase(s) (nSMase), ceramide, and the atypical protein kinase C (aPKC) and protein kinase M zeta (PKMζ) (Zhang et al. 2012, also see Nicol and Vasko 2007 for review). On the question of the relative importance of TrkA and p75NTR in NGF-induced hyperalgesia, controversial results have been reported, such as that NGF still produces hyperalgesia in p75 knockout mice (Bergmann et al. 1998, also see Lewin and Nykjaer 2014 for review).
8 Therapeutic Perspective
Efforts for the antagonization or reduction of NGF action have been directed toward the development of (1) humanized monoclonal antibodies (mAbs), (2) small molecules that bind NGF and change its molecular shape such that it can no longer bind to its receptor(s), (3) peptides that competitively bind TrkA or p75NTR receptors (Eibl et al. 2012), and (4) small molecules that block TrkA activities (Ghilardi et al. 2011). The specificity of mAbs is quite high, but it must be intravenously or intramuscularly injected. In addition, administration of mAbs entails the risk of immune reactions. In contrast, while small molecule inhibitors of kinase activity may not be as specific as mAbs or small molecules that bind to NGF and block binding to its receptor, they may have equal therapeutic potential. They can be orally administered, are less expensive to produce, and have greater flexibility in dosing. Except for mAbs, these agents are still in the preclinical stage. Clinical trials using a humanized anti-NGF antibody, tanezumab, have been conducted for low back pain (Kivitz et al. 2013), osteoarthritis (Sanga et al. 2013), and bone cancer pain, and outcomes have been good. However, because of a serious side effect (joint destruction), all clinical trials except for one on bone cancer pain were for a while suspended and now restarted. We hope that in the near future, some of these agents can be used for the treatment of pain.
References
Aloe L, Tuveri MA, Carcassi U, Levi-Montalcini R (1992) Nerve growth factor in the synovial fluid of patients with chronic arthritis. Arthritis Rheum 35:351–355PubMed
Amann R, Schuligoi R, Herzog G, Donnerer J (1996) Intraplantar injection of nerve growth factor into the rat hind paw: local edema and effects on thermal nociceptive threshold. Pain 64:323–329PubMed
Related posts:





Full access? Get Clinical Tree


